Болезнь альцгеймера, лечение в домашних условиях
Уже на протяжении многих лет ученые пытаются найти надежное лекарство от болезни Альцгеймера. К сожалению, на сегодняшний момент данное заболевание не поддается терапии, удалось лишь найти способы замедлить развитие старческой деменции у пожилых людей.
Уже при появлении первых симптомов недуга стоит готовиться к прогрессированию патологии.
На некоторое время развитие болезни можно приостановить при помощи медикаментозной терапии.

Симптомы заболевания
Для различных стадий болезни имеются свои симптомы.
На ранней стадии
Сначала начинает страдать память.
Справка! Нарушения могут быть кратковременными или продолжительными:
- Кратковременные провалы в памяти развиваются постепенно, со временем человек вынужден записывать свои планы в блокнот, чтобы в очередной раз не забыть что-то сделать.
- Некоторые пациенты пытаются скрывать свои появившиеся провалы в памяти.
- Обычно родственники начинают замечать появившуюся болезнь в тот момент, когда человек часто забывает значительные моменты в жизни или события.
Позже наступает прогрессирующая утрата долговременной памяти:
- Человек теперь уже забывает не только о своих планах и обещаниях, он начинает забывать названия предметов, имена людей и т.д.
- Постепенно ослабевают мыслительные способности.
- Человеку становится сложно сосредоточиться.
- Человек начинает страдать от депрессии. Часто это связано с появлением провалов в памяти и мнительностью пациента.
- Ощущается и повышенная тревожность.
- Такое состояние приводит к психозам, которые сменяются апатией, потерей интереса ко всему окружающему.
Каковы первые симптомы и признаки болезни Альцгеймера рассказывается на видео:
Прогрессирование патологии
На этой стадии человек уже с трудом ориентируется в современности. Он не в состоянии вспомнить дату и даже год, в котором пребывает.
Человек может заблудиться, выйдя на улицу. Такие больные становятся чересчур подозрительными, что доходит до паранойи. Им тяжело контролировать свои эмоции.
Поздний этап
На этом этапе заболевания человек уже с трудом контролирует физиологические процессы в организме. Больной не идет на контакт с окружающими.
Многие пациенты с трудом передвигаются, часто прикованы к инвалидной коляске.
Причины возникновения
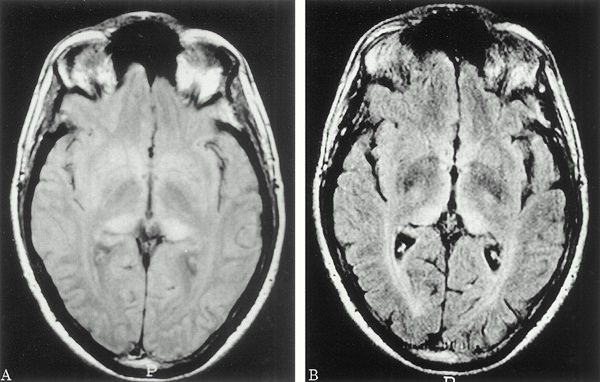
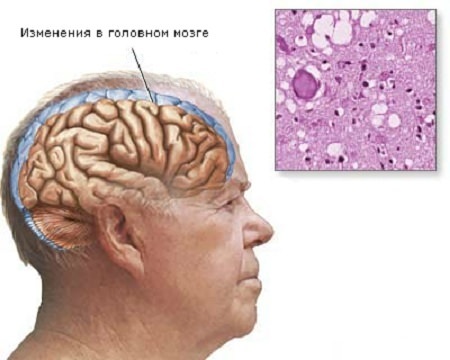
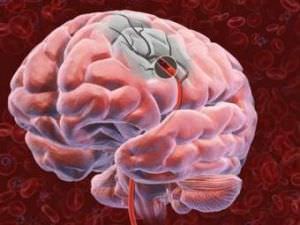
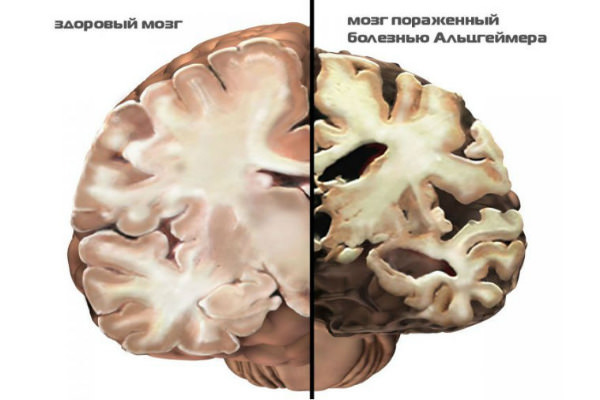
В настоящий момент точной причины развития болезни Альцгеймера не установлено. Существует мнение, что болезнь развивается из-за синильных бляшек, которые формируются в сосудах головного мозга, приводящих к отмиранию нейронов.
Стадии Альцгеймера и характерные признаки болезни
Другие возможные причины развития заболевания:
На видео доктор рассказывает о причинах возникновения болезни Альцгеймера:
Что происходит в организме?
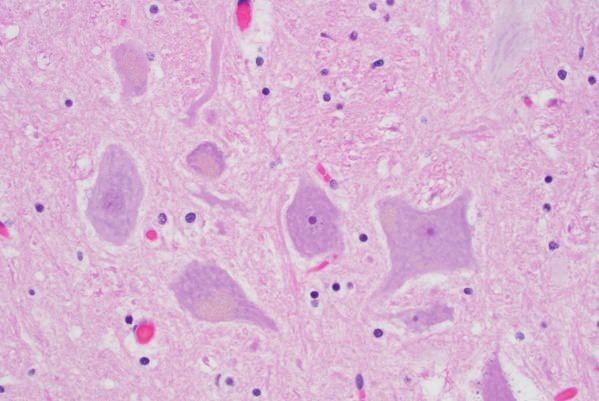
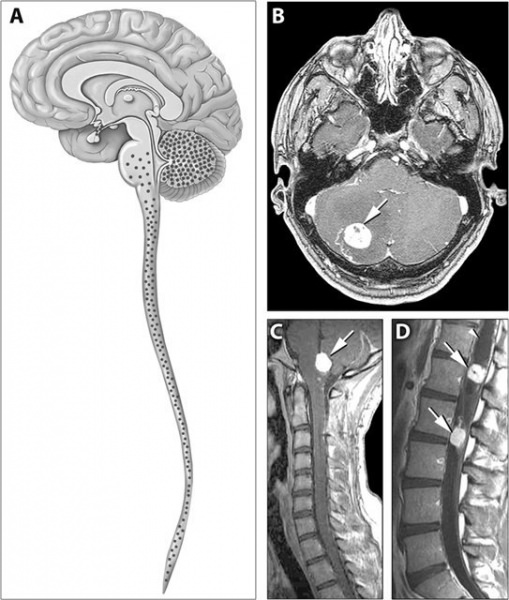
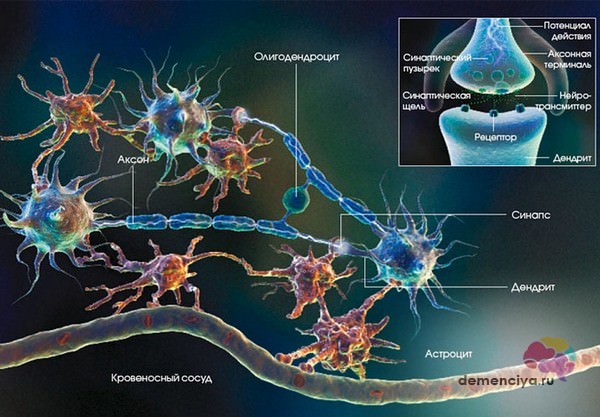
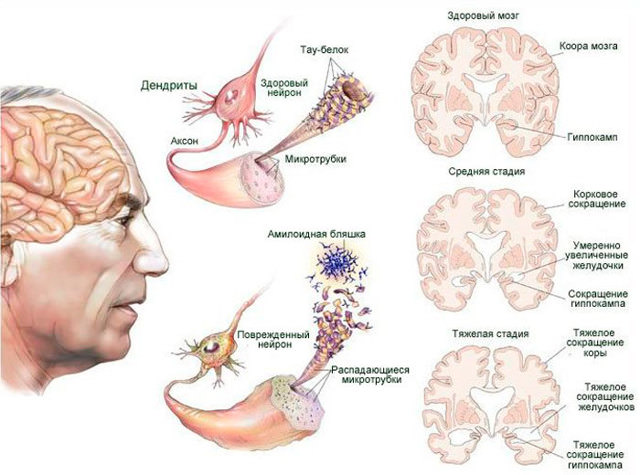
По мере развития болезни в мозговой части откладывается бета-амилоидный белок, из-за которого образуются бляшки в сосудах
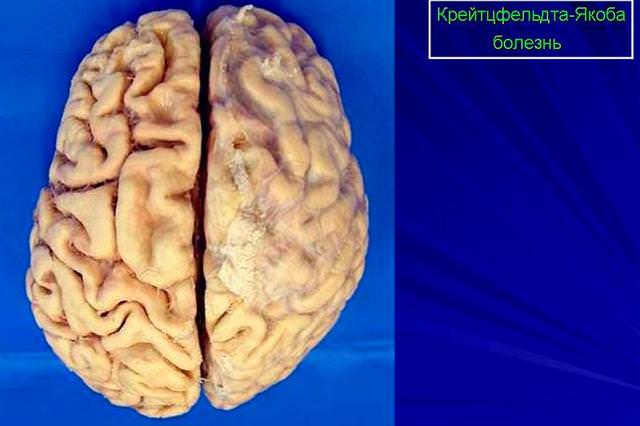
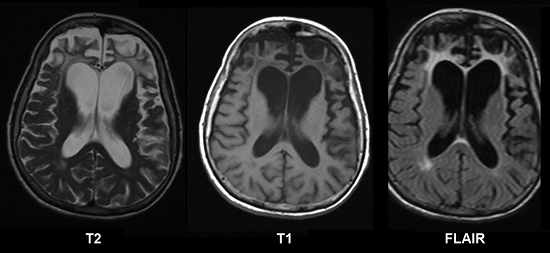
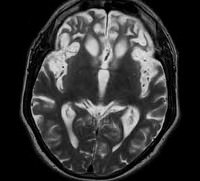
В результате повреждаются нейроны, образуя нейрофибриллярные клубочки. Все это нарушает работу связок между нейронами, а значит, приводит к сбою функций головного мозга (см. фото) .
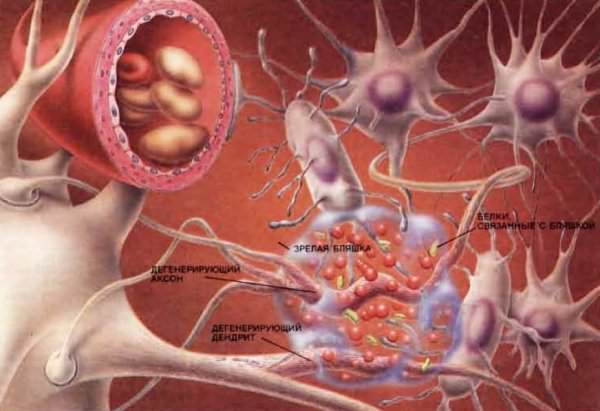
Причины отложения бета-амилоидного белка до конца не установлены.
В последние годы разработаны различные гипотезы относительно развития болезни Альцгеймера:
- Одна из них предполагает наличие особого гена, который способствует образованию бляшек из амилоидного бета-белка.
- По другим мнениям к причинам развития заболевания относятся воспалительные процессы и цитокины.
Можно ли вылечить?
Болезнь Альцгеймера относится к неизлечимым недугам, но имеются терапевтические меры, которые способны приостановить развитие патологии.
Справка! Лечение направлено в основном на снижение проявлений заболевания, например, оно помогает справиться с бессонницей, дезориентацией в пространстве, депрессивным состоянием, тревожностью и т.д.
Стадии заболевания
Существует несколько стадий данного заболевания.
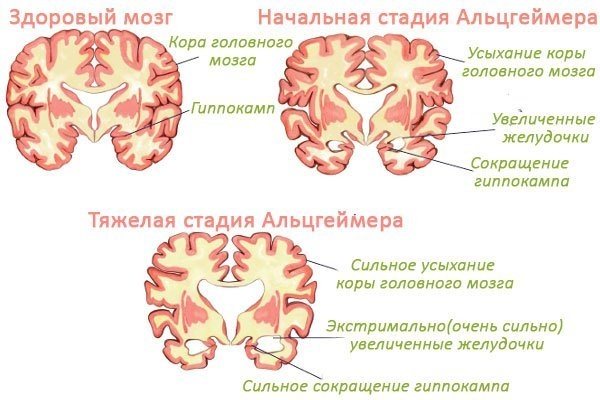
Начальная
Эта стадия делится в свою очередь на три подстадии:
Стадия фокальных расстройств
На данном этапе слабая деменция переходит в очаговую форму, при которой нарушаются определенные функции головного мозга:
Терминальная
Все расстройства, которые были перечислены выше, достигают своей максимальной стадии
Дополнтельно появляются следующие симптомы:
Три стадии болезни Альцгеймера описываются на видео:
Как лечить?
Полностью вылечить заболевание нельзя, но можно облегчить его симптомы.
Новое в лечении
В настоящий момент продолжают разрабатываться новые методики лечения данного заболевания. Вот некоторые новые препараты, которые появились сравнительно недавно.
Прививка от слабоумия
Этот препарат разработали специалисты из США. Основу вещества составляют компоненты, направленные на восстановление иммунитета человека.
В результате приема препарата происходит гибель патологических клеток-белков. Данный препарат находится на стадии разработки, в данный период к ученым из Штатов присоединились специалисты из Европы и Азии.
Таблетка в день и никаких патологий
Такое лекарство было разработано в Англии.
Его суть заключается в ежедневном приеме таблетки, которая блокирует и снижает уровень бета-амилоида в мозгу на 95 %.
Предварительно было проведено исследование на 200 добровольцах. Результат превзошел все ожидания, были явные показатели улучшения состояния.
Аэрозоль от потери памяти
Еще одно новшество в медицине, через дыхательные пути лекарственное вещество достигает мозга и способствует росту молодых клеток.
Даже если есть бляшки из амилоидных белков, вещество способно проникнуть через них и улучшить мозговую деятельность.
Стволовые клетки на страже мышления
Когда болезнь развивается, происходит отмирание клеток. Поэтому метод стволовых клеток предполагает их замену на новые и здоровые.
Если провести терапию правильно, то симптомы психологических расстройств полностью исчезнут. Разработали данный способ по замене мутировавших генов израильские специалисты.
Пластырь-новинка
Медикаментозные средства имеют массу побочных эффектов, противопоказаний и нередко вызывают аллергические реакции. Чтобы восполнить в организме нехватающие вещества, ученые предлагают использовать специальный пластырь.
В нем содержится определенная доза нужного лекарства, при этом он безопаснее своих таблетированных аналогов.
Тразодон
Применение народных средств в домашних условиях
С данной болезнью также борются с помощью народных средств.
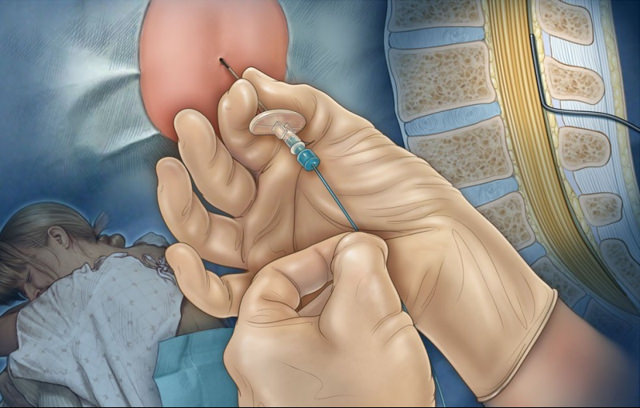
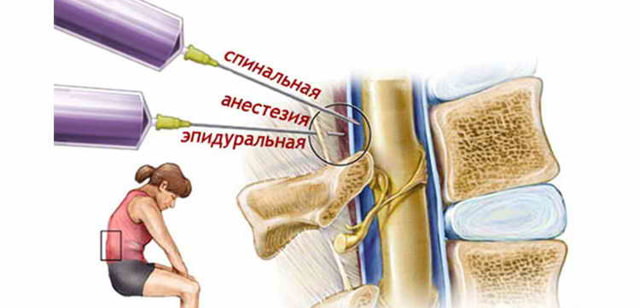
Диагностика болезни Альцгеймера – тесты, анализы
Диета
Определенная диета именно для этой болезни не разработана. Лечебное питание направлено на поддержание общего здоровья организма.

Для того чтобы укрепить стенки сосудов, улучшить их проходимость и отрегулировать обменные процессы, в рацион нужно включить следующие продукты:
Внимание! Исключить нужно сдобу, молочные продукты и сладости. Кроме того, приводит к дисфункции многих внутренних органов потребление специй, жирной, жареной пищи. Под запретом алкогольные напитки и курение.
- При соблюдение данных рекомендаций, человек зачастую начинает чувствовать себя намного лучше.
- Какой диеты стоит придерживаться при болезни Альцгеймера рассказывается на видео:
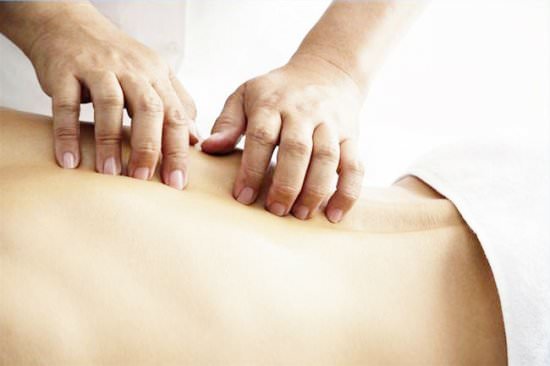
Массаж
Массаж направлен на устранение болезненности в некоторых частях тела, а также для лучшей циркуляции крови.
Первоначально все действия должен показать специалист, после этого данная обязанность может перейти одному из членов семьи, причем делать массаж нужно будет ежедневно:
Профилактика
Несмотря на то что профилактики от болезни Альцгеймера не существует, но все же можно попытаться предотвратить развитие этого заболевания следующими способами:
На видео рассказывается, как избежать болезни Альцгеймера:
К какому врачу обратиться?
В основном данной проблемой занимаются врачи-неврологи, также может понадобиться помощь и других узких специалистов – кардиолога, психиатра и т.д.
В Москве
В Москве множество клиник, специализирующихся на данных заболеваниях. Одна из них – клиника врача-психотерапевта И. Г. Гернета.
В Ростове
Клинико-диагностический центр «Медицина» в Ростове-на-Дону находится на улице Стрелковой Дивизии и в том числе занимается вопросами лечения болезни Альцгеймера.
В Новокузнецке
В Новокузнецке практикуют следующие врачи, занимающиеся лечением данного заболевания: Зиборова Светлана Станиславовна, Шарапова Ирина Николаевна в ГКБ №1.

В Израиле
В этой стране множество клиник по направлению психических расстройств человека, самые крупные из них:
Как и любое заболевание, болезнь Альцгеймера лучше предотвратить, чем лечить.
Для этого следует вести здоровый образ жизни, повышать свой интеллектуальный потенциал, есть правильную пищу и быть физически активным.
Болезнь Альцгеймера, лечение народными средствами

Болезнь Альцгеймера, лечение народными средствами
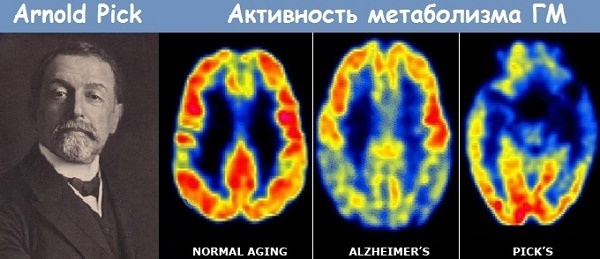
Болезнь Альцгеймера — это нейродегенеративное заболевание, самая распространенная форма деменции. Появилось оно еще в 1907 году, первый кто его диагностировал болезнь — Алоис Альцгеймер, известный немецкий психиатр, в честь которого и назвали заболевание.
Данная болезнь происходит вследствие отмирания нейронов мозга, что приводит к так называемому слабоумию.
Такое заболевание, как болезнь Альцгеймера, проявляется в пожилом возрасте после 65 лет, но первые признаки могут начать проявляться уже в сорок. Самую большую зону риска заболеваемости занимает население женского пола.
На сегодняшний день болезнь считается неизлечимой, еще не был создан препарат, способствующий излечить пациента.
Средний срок жизни с болезнью Альцгеймера, составляет порядка семи лет
Причины болезни
Существует ряд причин, судя из которых возникает тяжелое заболевание. Выдвинуто несколько гипотез возникновения, вот самые основные:
Причины возникновения болезни Альцгеймера
Развитие заболевания, полностью неизвестно
Причиной зачастую является генетическая предрасположенность. Бывает, что заболевание становиться семейным, и болезнь Альцгеймера передается от поколения к поколению.
Симптомы заболевания
Данное заболевание распознается по следующей симптоматике:
- Появляется нарушение памяти, которая усиливается, бывает кратковременная и полная потеря памяти;
- Затруднения в арифметических действиях, подсчет денег, расчет незамысловатого сложения или другого действия связанного с цифрами;
- Проблемы с быстрым принятием каких-либо решений;
- Утрата способности к написанию и чтению;
- Проявление паранойи и депрессии;
- Возникновение галлюцинаций;
- Потеря возможности узнавать родных и близких;
- Проблемы с туалетом, зачастую недержание;
- Неспособность к самообслуживанию.

Симптомы этапов болезни Альцгеймера
Диагностика заболевания
При возникновении одного из симптома, которые перечислены выше, следует обратиться к врачу, это неврологи и психиатры. Специалисты изучают историю заболевания, анамнез, задают наводящие вопросы для выяснения полной картины заболевания.
Это все из-за того, что болезнь Альцгеймера у каждого проявляется при наличии своих особенностей. Для диагностирования также применяют специально разработанные компьютерные тесты.
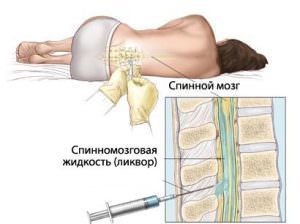
Существует целый ряд дополнительных методов, которые подтверждают диагноз.
Обычно проводят:
По окончанию процесса исследования, доктор составляет полную картину заболевания. После определения стадии, имеющегося заболевания, назначается необходимый курс лечения.
Выбор метода лечения
Существует ряд препаратов, которые позволяют существенно замедлить расширения заболевания, или же облегчить протекание болезни Альцгеймера. Медикаментозное лечение можно разделить по группам:
- Препараты, действие которых направленно на улучшение памяти, способствует улучшению мыслительного процесса, зачастую используют ривостигмин-содержащие медикаменты.
- Препараты, работа которых направлена на борьбу с психологическим состоянием, а именно апатия, чрезмерная тревога, нервозность, депрессия.
Все препараты требуют строгого назначения лечащим врачом, самолечение может сделать только значительно хуже.
Народные средства лечения
В лечении заболевания Альцгеймера дополнительно можно прибегнуть к народной медицине, но следует получить консультацию доктора, дабы не навредить. Стоит учитывать, что главное не переусердствовать с лечением народными средствами.

Народные средства
Такие средства имеют прямое усиление действия принимаемых препаратов, поддерживает на достаточном уровне физическое и психологическое состояния больного.
К средствам народного лечения относят не только травяные отвары, способствующие лечению, но и правильный рацион питания, физические упражнения и методы, активизирующие работу мозговой деятельности.
Рецепты из трав, настойки
В народных средствах лечения при болезни Альцгеймера зачастую применяют:
Рацион питания
Питаться больной должен качественной пищей, которая наполнена растительными жирами, обязательно в рацион должна входить рыба, фрукты, овощи. Одними из самых полезных считаются помидоры, лук, чеснок, черная смородина, апельсины.

Здоровый рацион питания
Очень важным моментом является наличие в продуктах антиоксидантов, а это: морковь, бобовые, капуста, сухофрукты. Для полноценной стимуляции нервов рекомендуется пить крепкие чаи.
Гимнастика
При болезни необходимо делать дыхательную гимнастику, прекрасно подойдут занятия йогой. Если человек уже очень пожилой, то необходимо совершать ежедневные пешие прогулки на свежем воздухе, можно с небольшими передышками.
Умственные нагрузки
При диагнозе болезнь Альцгеймера, необходимо напрягать умственные способности, разгадывать кроссворды, читать книги, учить произведения наизусть.

Умственные нагрузки
Данные действия будут способствовать умственной нагрузке.
Видео: Болезнь Альцгеймера
Заключение
Болезнь Альцгеймера и ее лечение, достаточно сложный момент, данное заболевание приводит к летальному исходу, следует относиться к своей нервной системе с осторожностью, в случае возникновения симптомов, как можно раньше обратиться к специалистам за помощью.
Лечение болезни Альцгеймера: медикаменты, народные средства, аппараты, логопед, психотерапия

В современном мире болезнь Альцгеймера представляет серьезную проблему и занимает четвертое место среди смертельных недугов.
Впервые описал заболевание знаменитый немецкий психиатр Алоис Альцгеймер, в честь него и назвали болезнь.
Чаще всего недуг поражает пациентов старше 60 лет. Лечением болезни занимаются психиатры и неврологи.
Каковы симптомы болезни Альцгеймера, можно ли вылечить заболевание, как и чем лечить недуг в домашних условиях, в чем заключается лечение?
Суть патологии

До сих пор не определена точная причина возникновения слабоумия. Общепризнанным является наследственный фактор.
Кроме этого, заболевание может возникнуть вследствие различных травм, воспалительных заболеваний мозга.
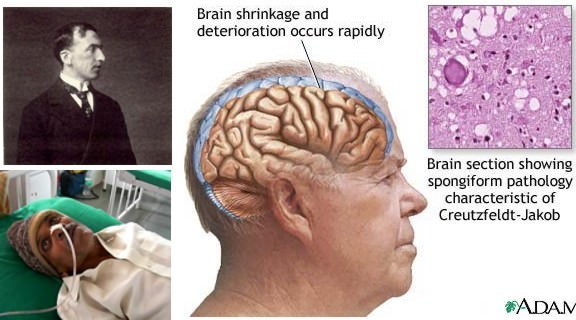
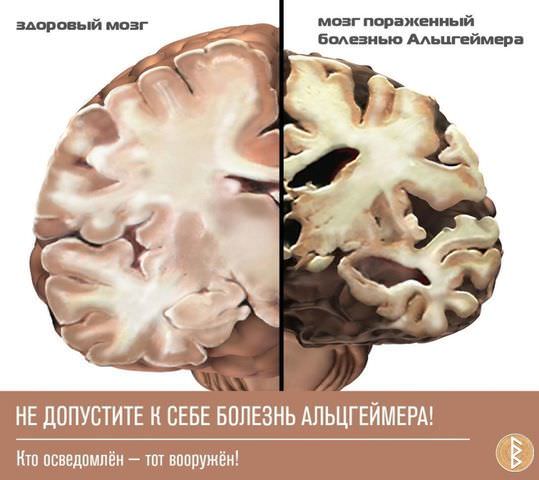
Недуг имеет органическую природу. Токсический белок (бета-амилод) откладывается между нейронами клеток головного мозга.
Появляются амилоидные бляшки, разрушающие связи между нейронами. В клетках образуются нейрофибрилляные клубочки, которые провоцируют полное отмирание клетки.
Еще один участник разрушающего процесса — липопротеин АроЕ.
К сожалению, на сегодняшний день не существует средств, способных вылечить деменцию полностью.
Есть препараты, которые разрушают нейрофибриллярные клубочки и препятствуют образованию бляшек. Однако, их эффективность слишком низкая.
- Кроме того, генные мутации не позволяют остановить разрушительный процесс.
- Поэтому лечение болезни Альцгеймера носит симптоматический характер, направлено на сохранение интеллектуальных навыков как можно дольше.
- Терапия позволяет оттянуть наступление последней стадии болезни, сохранить человеку способность обслуживать себя самому.
- О лечении болезни Альцгеймера расскажет видео:
К какому врачу обратиться, лечится заболевание или нет
Для успешного лечения заболевания проводится полная диагностика, чтобы определить стадию и распространение болезни.
Терапия включает в себя медикаментозное лечение и психологическую помощь.
Какой врач лечит болезнь Альцгеймера? Лечением занимаются неврологи и психиатры.
Кроме того, необходимо корректировать сопутствующие патологии (болезни сердца, щитовидки), от этого зависит положительный исход лечения.
Медикаменты
Сложности этого метода в том, что организм пожилого пациента ослаблен и слишком восприимчив к лекарствам. Поэтому назначать препараты нужно с осторожностью, начиная с маленьких доз, постепенно корректируя схему лечения.
В психиатрии препараты для лечения болезни Альцгеймера делятся на следующие группы:
- Галантамин. Оказывает влияние на повышение внимания и памяти. Обладает довольно слабой токсичностью, хорошо переносится больными;
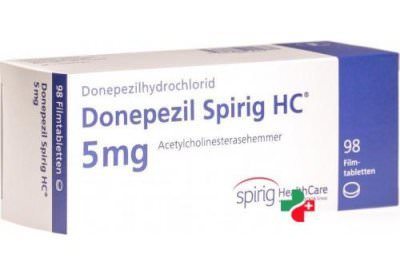
- Донепезил. Считается наиболее эффективным среди всех препаратов. Скорость ингибирования холинэстеразы повышается в 1000 раз быстрее, чем при использовании других лекарств;
- Ревастигмин. Применяется при сильно прогрессирующем течении болезни.
- Оланзапин;
- Клозепин;
- Рисперидон;
- однако, при острых психических состояниях (галлюцинации, бред) применяют Галоперидол.
Традиционно применяют Церебролизин и Актовегин. Эти средства улучшают мозговое кровообращение, способствуют снабжению клеток мозга кислородом.
Эффективность лечения достигается за счет длительного применения препаратов. Первые улучшения можно заметить по прошествии нескольких недель.
Среди новых методов лечения — применение стволовых клеток. Поврежденные клетки замещаются здоровыми, функции мозга восстанавливаются. Однако, этот метод является довольно дорогим и еще до конца не изученным.
Ингибиторы ацетилхолина (Донепезил), Мемантин и успокоительные при болезни Альцгеймера:
Народные средства
Применение народных методов оправдано в сочетании с медицинскими препаратами. Как моносредство они бесполезны.
Какими травами хорошо лечить болезнь Альцгеймера? На начальном этапе средства народной медицины могут слегка замедлить прогрессирование патологии:

Из витаминов принимают добавку — соевый лецитин. Он улучшает работу головного мозга.
Кроме того, полезно употреблять крепкий чай и кофе. Также хороший эффект дает постоянный прием Омега-3.
Важно! Доказано, что употребление трех чашек натурального кофе в день снижает риск деменции в несколько раз.
Кофе и болезнь Альцгеймера:
Какие аппараты применяются
Как еще бороться с болезнью Альцгеймера? В настоящее время предпринимаются попытки найти эффективные и безопасные методы борьбы с недугом.

Не так давно на рынке появился аппарат «Биомедис М». Это биорезонансный аппарат, излучающий электромагнитные волны высокой частоты. Они воздействуют на мозг и стимулируют его работоспособность.
Однако, этот метод является спорным. Как самостоятельное средство его применять не рекомендуют, так как деменция является следствием органического поражения клеток, восстановить разрушительный процесс данному аппарату не под силу.
Помощь психологов, логопедов
Применение психотерапевтических методик на начальном этапе значительно облегчает состояние больного и тормозит процесс развития болезни Альцгеймера.
Психологическая помощь оказывается в виде:
- арттерапии;
- лечения музыкой;
- обеспечения присутствия.
Выполнение различных умственных упражнений (решение задач, разучивание стихов, разгадывание кроссвордов, чтение книг).
Поможет ли логопед при болезни Альцгеймера? Да, при нарушении речи понадобится помощь логопеда.
Большое значение имеет психологическая поддержка близких. Важно хвалить больного за любые достижения, подбадривать.

Нельзя изолировать человека от общения, он должен посещать кружки, ходить в кино. Обязательны прогулки на свежем воздухе.
Родственникам нужно смириться с тем, что близкий человек уже никогда не стане прежним. Нельзя раздражаться, обижаться на него.
Если больной уже не может самостоятельно себя обслуживать, это придется делать близким, либо прибегнуть к помощи сиделки.
Болезнь Альцгеймера — необратимый процесс. Она неизбежно будет прогрессировать. При отсутствии адекватного лечения деградация личности и смерть больного произойдет намного быстрее.
Излечима ли болезнь Альцгеймера? Полностью вылечить таких больных современная медицина не в состоянии.
Лечение направлено на продление начальной стадии, сохранение интеллектуальных навыков на как можно более долгий период.
Эффективная терапия сочетает в себе применение современных медицинских препаратов, средств народной медицины и методов психологической помощи.
Успех лечения зависит от правильно подобранных комплексных методик.
Как лечить болезнь Альцгеймера в домашних условиях народными средствами
Болезнь Альцгеймера — тяжелейшее заболевание, поражающее человеческий организм в пожилом возрасте. Ученые до сих пор не пришли к однозначному выводу: каковы причины развития патологии.
В настоящее время терапия заболевания дает возможность лишь приостановить разрушительные процессы, протекающие в мозговых клетках, она включает в себя курсовой прием лекарственных препаратов и использование методов народной медицины.
Как лечить болезнь Альцгеймера в домашних условиях, какие народные средства помогут существенно улучшить качество жизни пожилого человека?
Целебные отвары из трав — вспомогательные средства, способные усилить действие лекарств, назначаемых при заболевании. Физические упражнения, правильный режим питания и методики, повышающие активность работы мозговых клеток — вот методы помочь больному в домашних условиях.
Особенности лечения в домашних условиях
К особенностям терапии болезни Альцгеймера необходимо отнести неукоснительное соблюдение режима дня. Ежедневно следует принимать назначенные лекарства, поддерживать организм с помощью народных средств, заниматься физическими упражнениями и тренировкой мозга.
Двигательная активность зависит от возраста пациента. Лечащий доктор назначает комплекс физических упражнений для каждой возрастной группы.
Пожилому человеку с таким диагнозом следует больше времени проводить на свежем воздухе, а выполнение упражнений можно заменить на неспешную прогулку, которая длится не менее часа.
Для тренировки мозга необходимо ежедневно заучивать небольшие отрывки стихотворений или прозы, собирать пазлы, заниматься разгадыванием сканвордов и кроссвордов.
Фитотерапия
Лечение и профилактика Альцгеймера народными средствами включает в себя прием отваров из лекарственных растений, являющимися натуральными антидепрессантами, способствующими усилению кровотока, стимулирующими работу мозга.

Отвары и настои, которые используют при заболевании:
- Настой из плодов лимонника и корня женьшеня.
Сушеный корень женьшеня измельчить, плоды лимонника раздавить тупой стороной ножа, взять равные пропорции сырья, смешать. Чайную ложку смеси поместить в термос с широким горлом, залить одним литром кипятка, настоять пару часов. Принимать настой в небольшом количестве несколько раз за день. Настой положительно влияет на активность мозговых клеток.
- Отвар из пустырника обыкновенного.
Столовую ложку сушеного сырья поместить в стеклянную посуду, залить двумя стаканами горячей воды, варить на паровой бане 9-12 минут, остудить, процедить, добавить еще 2 стакана кипяченой воды. Пить отвар 2-3 раза за день по полстакана. Средство поможет нормализовать показатели АД, снять агрессию, повышенную возбудимость.
- Целебный чай из зверобоя и вереска.
Сушеные листья растений добавлять в заварку, пить целебный чай в течение всего дня. Растения относятся к сильным антидепрессантам.
- Настой из листьев гинкго билоба.
Сушеные листья растения, в количестве пятидесяти грамм, помещают в бутылку темного стекла и заливают двумя стаканами водки, дают настояться в течение двух недель. Настой процеживают и принимают в небольших количествах ежедневно. Точная дозировка и длительность приема должна быть назначена лечащим врачом.
- Настой из диоскореи кавказской.
Корни растения тщательно промывают, измельчают, помещают в темную бутылку, заливают водкой из расчета: 4 стакана водки на 100 грамм сырья. Настаивают в темном помещении не менее двух недель, процеживают, пьют одну чайную ложечку по 3 раза за день, перед приемом пищи. Средство поможет стимулировать работу головного мозга.

Лечение в домашних условиях болезни Альцгеймера включает в себя курсовой прием отваров из аира, полыни, цикория, девясила, эхинацеи, элеутерококка и аралии. Эти растения повышают защитные свойства иммунной системы, придают тонус, улучшают работу головного мозга.
Продукты пчеловодства
Болезнь Альцгеймера лечение народными средствами включает в себя и ежедневное употребление продуктов пчеловодства.
Натуральный мед, перга, маточное молочко и цветочная пыльца — природные антиоксиданты, способные в разы уменьшить склеротические явления, улучшить кровоток, придать тонус стенкам сосудов.
Больному с таким заболеванием рекомендовано ежедневно употреблять в пищу 2-3 ложки сладкого лекарства. Медом можно заменить сахар, который негативно влияет на организм больного.
Продукты питания
В рационе питания больного должны присутствовать следующие продукты:
Питьевой режим
Питьевой режим чрезвычайно важен для пациента с таким диагнозом. Недостаток жидкости в организме может привести к ухудшению состояния.
За день необходимо выпивать не менее 1,5 литров жидкости (вода, соки, компоты из свежих ягод и сухофруктов).
Многие врачи сходятся во мнении, что именно систематический недостаток жидкости в организме и вредные привычки (алкоголь, никотин) являются факторами, провоцирующими развитие заболевания.

Один из способов стимулировать работу коры головного мозга — ежедневное употребление нескольких чашек свежезаваренного чая (черного или зеленого).
Другие средства
Для лечения заболевания в домашних условиях врачи часто рекомендуют:
- занятия йогой;
- дыхательную гимнастику;
- гирудотерапию (лечение пиявками);
- курсы расслабляющего массажа с маслами (кокосовое, персиковое).
Наука не стоит на месте, ежедневно появляются новые средства, способные улучшить состояние больного с диагнозом «болезнь Альцгеймера».
Болезнь Альцгеймера: лечение в домашних условиях – эффективно ли? Современные методы лечения в клинике Москвы
Болезнь Альцгеймера – это нейродегенеративное заболевание, характеризующееся поражением клеток головного мозга, расстройством функций мозга, развитием старческого слабоумия.
Заболевание протекает с необратимыми изменениями в мозге, имеет несколько стадий развития, заканчивается смертью больного после утраты организмом всех функций.
Болезнь Альцгеймера диагностируется в большинстве случаев у людей старше 60 лет, в редких случаях в более молодом возрасте.

Болезнь Альцгеймера, обнаруженная в молодом возрасте, быстро прогрессирует, заканчивается летальным исходом через 3-6 лет после диагностики заболевания. Данная форма болезни чаще всего имеет наследственную причину.
Заболевание у людей старшего возраста протекает медленно, продолжительность жизни зависит от своевременного назначения адекватного лечения.
До настоящего времени неизвестны настоящие причины развития и ход течения заболевания, а лишь факторы, которые приводят к развитию патологии.
При появлении первых признаков заболевания следует записаться на прием к неврологу Юсуповской больнице, который подберет оптимальную терапию.
Лечение болезни Альцгеймера народными средствами
Лечение заболевания народными средствами без консультации специалиста недопустимо.
Заболевание имеет необратимый характер, если не принять все меры своевременно, оно быстро прогрессирует и может привести к летальному исходу.
Средняя продолжительность жизни больного Альцгеймером от момента диагностики около 8 лет. В большинстве случаев диагностируется заболевание при появлении выраженных симптомов, на умеренной (средней) стадии болезни.
Народные средства при лечении Альцгеймера используют для снятия симптомов заболевания. Применение трав способствует снижению возбудимости больного, артериального давления, прием определенного состава трав стимулирует иммунитет. Лечение народными средствами должно быть согласовано с лечащим врачом.
Для улучшения состояния больного применяется диета, разработанная специалистом диетологом. Доказано, что развитию заболевания способствует недостаток витаминов группы В.
Питание больного должно содержать витамины группы В, рекомендованы омега-3 жирные кислоты, виноградный сок или изредка немного красного вина (одни из лучших антиоксидантов).
Рацион больного должен содержать достаточное количество овощей и фруктов, в питании должны присутствовать орехи, молоко и кисломолочные продукты, зелень, яйца, злаки, нежирное мясо, печень, дрожжи.
Нельзя давать больному жирное мясо, мучные изделия, острые приправы, алкоголь, следует заменить сахар мёдом. Больной должен пить достаточное количество жидкости.
Какое лечение эффективно при болезни Альцгеймера
Все известные методы лечения направлены на смягчение существующих симптомов. Современные препараты, способные приостановить течение заболевания, находятся на этапе исследований и разработок.
Обнаружено позитивное влияние инновационных препаратов на ранней стадии развития заболевания, на более поздних стадиях болезни практически все препараты оказались бесперспективными.
На этих стадиях заболевания врачи применяют лечение, направленное на смягчение симптомов.
Для поддержания работы мозга в нормальном состоянии требуется применять следующие методы:
- гимнастику. Спортивные занятия помогут больному на раннем этапе болезни, будут стимулировать мышцы, оказывать влияние на работу мозга. Больным на поздних стадиях заболевания показан массаж;
- очень полезны музыкальные занятия, цветотерапия, ароматерапия. Такие занятия воздействуют на рецепторы, активизируют работу мозга;
- для улучшения работы мозга используют умственные нагрузки. Больному полезно играть в головоломки, отгадывать кроссворды, освоить компьютер;
- ежедневные прогулки в парке, в тихом месте помогут поддержать эмоциональное состояние больного в равновесии. Стресс и хроническая депрессия способствуют развитию болезни Альцгеймера;
- обращение к психологу позволит скорректировать психологические проблемы.
Лечение болезни Альцгеймера в Юсуповской больнице
Как заниматься с больным и правильно ухаживать, вы можете узнать у специалиста Юсуповской больницы. При появлении первых признаков заболевания следует записаться на прием к неврологу.
Лечение заболевания в домашних условиях будет тяжелым испытанием для всех членов семьи. Самым трудным этапом будет забота о больном с последней стадией болезни Альцгеймера.
Помощь психолога также требуется и членам семьи заболевшего.
В Юсуповской больнице для диагностики болезни Альцгеймера используют инновационную аппаратуру известных мировых производителей. Опыт неврологов больницы позволяет дифференцировать болезнь Альцгеймера от других заболеваний и назначить адекватное лечение.
Обращение на поздних стадиях заболевания не дает возможности эффективно воздействовать на патологию, так как в мозге произошли тяжелые, необратимые изменения, поражены большие участки тканей мозга. Записаться на консультацию к неврологу вы сможете по телефону.
- МКБ-10 (Международная классификация болезней)
- Юсуповская больница
- Ю. Г. Каминский, Е. А. Косенко «Популярно и не очень о болезни Альцгеймера» Либроком, 2009 г., 136 стр.
- Билл Грант «Старческое слабоумие. Болезнь Альцгеймера и другие формы.» Alzheimer’s Disease. A Carer’s Guide Серия: Советы врача Норинт, 2003 г., 80 стр.
- Cummings JL, Frank JC, Cherry D, Kohatsu ND, Kemp B, Hewett L, Mittman B (2002). «Guidelines for managing Alzheimer's disease: Part II. Treatment». American Family Physician 65 (12): 2525–2534.
*Информация на сайте носит исключительно ознакомительный характер. Все материалы и цены, размещенные на сайте, не являются публичной офертой, определяемой положениями ст. 437 ГК РФ. Для получения точной информации обратитесь к сотрудникам клиники или посетите нашу клинику.
Скачать прайс на услуги

Как можно лечить болезнь Альцгеймера в домашних условиях народными средствами?
Конечно же, о том, как лечить болезнь Альцгеймера в домашних условиях, в первую очередь должны знать родственники людей, которые страдают от столь распространенного недуга. При наличии данного заболевания поражению постепенно подвергаются практически все нервные клетки мозга, что вызывает их дегенерацию.
Этот процесс способствует тому, что мозг утрачивает способность нормально анализировать и применять ту или иную информацию. Практически во всех случаях дегенерация мозговых клеток приводит к тому, что человек теряет свою самостоятельность, память и перестает нормально ориентироваться в окружающей обстановке.
Страдают от данной болезни люди старше 65 лет, однако иногда можно отыскать пациентов, которым исполнилось всего лишь 50.
Перед тем как провести лечение в домашних условиях, нужно получить наиболее полную информацию о симптомах заболевания и причинах его возникновения. К примеру, среди самых распространенных предвестников развития болезни можно выделить:

- депрессию;
- потерю чувства времени;
- перепады настроения;
- нарушение памяти;
- беспокойство.
Нужно помнить, что человеку, у которого абсолютно внезапно появилось нарушение памяти и в семье есть случаи развития болезни Альцгеймера, необходимо сразу же обратиться к квалифицированному медику для установки диагноза. Это поможет намного быстрее начать лечение.
На сегодняшний день в медицине нет каких-либо точных данных, которые смогли бы рассказать о главной причине развития болезни. Некоторые медики уверенны, что это может быть дефицит эстрогенов, сосудистые мозговые приступы, частые стрессы, генетическая предрасположенность, а также недостаток в ежедневном рационе питания жирных кислот.