Болезнь крейтцфельдта-якоба или синдром кортико-стриоспинальной дегенерации
Болезнь Крейтцфельдта-Якоба (БКЯ) или как ее еще называют синдром кортико-стриоспинальной дегенерации относиться к патологиям с быстропрогрессирующим течением. Для нее свойственны дистрофические изменения тканей головного мозга.
Такое явление обычно ведет к развитию деменции (слабоумию) и скорому летальному исходу. Симптоматика патологии фактически идентична альцгеймеровским проявлениям.
Однако, в отличие от них, болезнь Крейтцфельдта-Якоба значительно быстрее развивается.
Такой вид патологии может быть классическим, то есть вызываться спонтанно или являться следствием генных мутаций и приобретенным. Ко второму типу относится заражение от больного животного или человека, а также употребление инфицированной говядины.
Последний случай был выявлен в 1986 году после вспышки эпидемии коровьего бешенства (нейродегенеративной прионной болезни) в Англии. С тех пор болезнь Крейтцфельдта-Якоба считается одним из проявлений прионного заболевания. Она также называется губчатым энцефалитом из-за внешнего вида измененных тканей головного мозга.
Возникают такие нарушения из-за воздействия прионов, которые являются инфекционными агентами.
Губчатый энцефалит проявляется крайне редко как в классическом спонтанном варианте, так и вследствие коровьего бешенства. По статистике ежегодно лишь в 1 случае на 1 миллион человек населения ставится такой диагноз и в основном у возрастных людей.

Причины
Губкообразная энцефалопатия, вызываемая болезнью, является следствием воздействия патогенных белков (протеинов), которые называются прионами. В норме они абсолютно безвредны, но вследствие влияния определенных факторов протеины подвергаются изменениям. Понять, как возникает синдром Крейтцфельдта-Якоба можно, взглянув на ее виды:
- Спонтанное проявление. Такой тип возникновения синдрома кортико-стриоспинальной дегенерации относится к классическому и не имеет явных причин появления;
- Генетическая аномалия. В Америке губкообразная энцефалопатия в 10% случаев возникает из-за генной мутации. Такая разновидность называется семейной болезнью Крейтцфельдта-Якоба и передается преимущественно по наследству;
- Заражение. В редких случаях синдром кортико-стриоспинальной дегенерации проявляется у людей после попадания в кровь инфицированных частичек, полученных от больного человека или животного. Такое явление возможно после пересадок кожного покрова или других тканей. Иногда заражения происходит вследствие оперативного вмешательства на головном мозге. В этом случае передача идет с поверхности инструментов, так как нельзя убить прионы обычными методами стерилизации. Заразиться от животного также возможно, например, из-за приема лекарственных средств, полученных из его головного мозга. В этом случае речь идет о синдроме ятрогенного типа;
- Коровье бешенство. Случаи распространения такой разновидности болезни крайне редки. Подхватить ее можно, употребляя плохо приготовленную говядину или работая с сырым инфицированным мясом. Этот вид патологии называется коровья губчатая энцефалопатия.
Группы риска
Большинство случаев синдрома кортико-стриоспинальной дегенерации являются спонтанными и установить причины их развития не представляется возможным. Однако для этого патологического процесса свойственны определенные факторы, влияющие на его развитие, а именно:
- Возраст больного. Обычно к 60-65 годам возникает спонтанный тип болезни, а от семейной разновидности люди страдают после 45-50 лет. Заражение болезнью Крейтцфельдта-Якоба и вовсе не имеет возрастных рамок. Для примера во время крупной эпидемии коровьего бешенства в Англии большинству заболевших было меньше 35-40 лет;
- Генетическая предрасположенность. Такой фактор обычно касается семейной разновидности синдрома кортико-стриоспинальной дегенерации. Возникает он вследствие генной мутации и передается по аутосомно-доминантному типу. Такой механизм распространение означает то, что достаточно наличия генного дефекта у одного из родителей и при этом будет около 50% вероятности рождения ребенка с этой аномалией. Однако наследственная предрасположенность также влияет и на другие формы патологии. У больных с ятрогенным БКЯ и коровьей губчатой энцефалопатией чаще всего имеются дефектные гены. Такое явление свидетельствует о том, что генетическая предрасположенность увеличивает шансы заразиться прионной патологией от больного человека или животного;
- Влияние инфицированного объекта. Пересадка тканей от инфицированного животного или человека, страдающего от БКЯ, способна воздействовать на развитие патологического процесса. Особенно если у больного есть генные аномалии.
Сегодня подхватить коровье бешенство крайне сложно, так как ее вспышки возникают редко и преимущественно в слаборазвитых странах. Для примера заразиться этой патологией в Англии почти нереально. Связано это с тщательным контролем и лишь в 1 блюде на 10 миллиардов можно встретить зараженную говядину.
Симптомы
Развивается БКЯ молниеносно и явные признаки деменции становятся заметны уже через несколько месяцев. Среди ее базовых проявлений можно выделить следующие:
- Спонтанные и быстрые движения;
- Проблемы с глотанием;
- Изменение поведения;
- Чувство тревоги;
- Депрессионное состояние;
- Ухудшение памяти;
- Снижение умственных способностей;
- Ухудшение зрения;
- Нарушение ритма сна;
- Возникновение речевых дефектов.
Для болезни Крейтцфельдта-Якоба свойственно быстрое прогрессирования, поэтому в считаные месяцы состояние больного значительно ухудшается. Большинство людей впадает в коматозное состояние. Летальный исход часто случается из-за дыхательной или сердечной недостаточности, а также вследствие инфекций, способных вызвать пневмонию и другие похожие патологии. Прогноз крайне неутешителен.
В лучшем случае человек может прожить с этим заболеванием около 2 лет, а в худшем лишь 3-4 месяца.
Если патологический процесс является следствием употребление инфицированной говядины, то симптомы расстройства психики проявляются не так очевидно. Нарушение когнитивных функций и вовсе возникает на более поздних сроках. Однако исход этой разновидности болезни аналогичен классическому типу БКЯ. Люди умирают примерно через 2 года от начала развития патологического процесса.
Диагностика
После выявления первых признаков заболевания, необходимо немедленно отправиться в больницу. Узнать о том, есть ли синдром кортико-стриоспинальной дегенерации у человека или нет можно, но лишь с помощью взятия на анализ ткани головного мозга (биопсии) или посмертно. В большинстве случаев врачу остается лишь предполагать наличие патологии у пациента. Для этого он смотрит на семейный анамнез, возникающую симптоматику и на результаты инструментального исследования:
- Электроэнцефалография. Ее суть заключается в измерении электрической мозговой активности;
- Магнитно-резонансная томография. С ее помощью врач может детально рассмотреть серое и белое мозговое вещество и выявить патологические отклонения, в том числе и дистрофического характера;
- Люмбальная пункция. Такую процедуру проводят для изъятия на анализ ликвора (спинномозговой жидкости);
- Биопсия миндалин. По ее составу врач может судить о наличии в организме инфекции, вызывающей коровье бешенство, но такой метод диагностики менее точный, чем остальные.
По результатам обследования невропатолог может выявить нарушения свойственные болезни Крейтцфельдта-Якоба. Однако точно узнать о наличии патогенных прионов в организме можно лишь с помощью биопсии тканей головного мозга.
Курс терапии
На сегодняшний день так и не было придумано ни одного действующего лекарства против БКЯ. За несколько десятков лет ученые не смогли сдвинуться в этом вопросе, и любая форма такой патологии со временем приводит к летальному исходу. Единственное чем может помочь современная медицина, так это облегчить общее состояние больного с помощью симптоматической терапии.
Осложнения
Синдром кортико-стриоспинальной дегенерации может привести к смерти за полгода, поэтому считается крайне опасным. Больные уже в первые месяцы начинают вести себя странно, отталкивают близких людей и даже перестают их узнавать. Постепенно они забывают о том, что нужно мыться, спать, есть и т. д.

Среди физических осложнений, которые могут привести к летальному исходу можно выделить следующие:
- Сердечная и дыхательная недостаточность;
- Развитие инфекционных заболеваний.
Болезнь Крейтцфельдта-Якоба является смертельным приговором, от которого нет лечения. Однако ученые продолжают вести исследования, цель которых остановить развитие патологии, поэтому больным не нужно отчаиваться и стараться продлевать свою жизнь и дальше. В таком случае всегда остается шанс дождаться желаемого лекарства.
Болезнь Крейтцфельда-Якоба, как основное проявление губчатой энцефалопатии
Болезнью Кройцфельдта-Якоба, (БКЯ, трансмиссивной спонгиоформной энцефалопатией, коровьим бешенством синдромом кортикостриоспинальной дегенерации) принято называть прогрессирующее дистрофическое поражение базальных ганглиев, коры большого и спинного мозга.
Это важно! БКЯ можно отнести к основным проявлениям прионной болезни (губчатой энцефалопатии).
Информация о эпидемиологии и патогенезе
Данное заболевание представляет собой около 85 % всех губчатых энцефалопатий, которое поражает людей всех рас и национальностей, женщин и мужчин, взрослых и детей. В большинстве случаев начало болезни приходится на средний или поздний возраст, но может развиться в любом возрасте.
КБЯ можно отнести к группе медленных вирусных инфекций. Определенную роль в развитии патологии играет генетическая предрасположенность (частые случаи семейной заболеваемости).
- Если говорить о патогенезе, то на сегодняшний день он не известен.
- Имеются сведения о снижении активности ряда ферментов (ДРН-липоамид-дегидрогеназа и лактатдегидрогеказа) и в коре головного мозга, это говорит о нарушении окислительных процессов.
- Наблюдается общее снижение количества липидов в сером веществе.
Это важно! Причина болезни Крейтцфельдта-Якоба и прочих губчатых энцефалопатий – аномальные белки, которые называются прионами.
Прионами называется инфекционные агенты особого класса, которые представляют собой белки с аномальной трехмерной структурой, в которых отсутствуют нуклеиновые кислоты.
Прионы не относятся к живым организмам, но размножаются за счет функций живых клеток (в связи с этим они имеют схожесть с вирусами).
Прионы — белки с аномальной третичной структурой, обладающие способностью ускорять конформационное превращение схожих нормальных клеточных белков в себе подобные.
При попадании в организм, прионы оседают на поверхности клеток, в результате взаимодействия нормальных белков с прионами, структура первых изменяется на патологическую.
На фоне скопления патологических беков на поверхности клеток происходит блокировка мембранных процессов — запускается процесс апоптоза (запрограммированная смерть клеток).
Какие существуют пути заражения БКЯ?
Это важно! Риск заражения данной болезнью очень мал. Прионы не передаются воздушно-капельным путем (кашель, чихание), а также половым путем. Но в сером веществе больных людей и животных содержатся прионы.
Принято различать следующие три пути возникновения БКЯ:
Но в большинстве случаев болезнь возникает спонтанно по непонятным причинам, поэтому точно установить факторы риска не всегда возможно.

Клиническая картина трансмиссивной спонгиоформной энцефалопатии
Клиническими критериями деменции при наличии БКЯ считаются:
- быстро прогрессирующая деменция с отсутствием всех высших корковых функций;
- дизартрия;
- Хореотетоз (экстрапирамидные нарушения);
- пирамидные нарушения (спастический парез);
- эпилептические припадки;
- миоклонус;
- Диплопия (зрительные нарушения);
- атаксия, акинетический мутизм.
Если говорить о продолжительности заболевания, то она не превышает 1− 2 лет.
Принято различать следующие стадии данного синдрома:
- Продромальный период — симптоматика неспецифична и возникает примерно у 30 % больных, появляется за недели и месяцы до первых признаков деменции (астения, нарушение сна, внимания, аппетита, мышления, памяти, потеря веса, поведенческие изменения, снижение либидо).
- Инициальный период — наличие головных болей, зрительных нарушений, головокружение, парестезии и неустойчивость и парестезии.
- Развернутый период — спастический прогрессирующий паралич конечностей с наличием экстрапирамидных признаков (тремор, ригидность и характерные движениями). Возникает атаксия, атрофия верхнего двигательного нейрона, снижение зрения и мышечная фибрилляция.
Диагностические мероприятия
Заболевание должно предполагаться во всех случаях деменций с быстрым прогрессированием в течение нескольких месяцев или лет, которые сопровождаются наличием множественных неврологических симптомов.
Квалифицированные специалисты могут предположить вероятность БКЯ, основываясь на данных семейного анамнеза, неврологических признаках, а также основываясь на результатах нескольких параклинических способов диагностики:
- результат тестирования когнитивных функций (меньше 24 баллов по шкале MMSE);
- МРТ;
- Позитронно-эмиссионная томография;
- Люмбальная пункция (анализ ликвора- анализ давления, цитоз, уровень сахара, наличие криптококкового антигена, вирусных агентов и бактериальных культур. Важный диагностический критерий – уровень маркера в ликворе;
- прижизненная биопсия мозга (при наличии проинформированного согласия опекунов, родственников или в случае, если пациент недееспособен);
- гистологическое и морфологическое исследование тканей головного мозга (подкорковых ядер, коры) при посмертной диагностике (аутопсии).
Это важно! Только с помощью биопсии мозга или посмертного осмотра мозговой ткани можно однозначно установить БКЯ.
Лечение губчатой энцефалопатии
На сегодняшний день этиотропная терапия не разработана. Показано симптоматическое лечение для уменьшения симптоматики.
Это важно! При наличии БКЯ следует необходимо отменить прием всех медицинских препаратов, которые могут оказать негативное воздействие на поведение пациента и мнестические функции.
Что касается медикаментозного лечения, то традиционные противовирусные препараты (интерфероны, амантадин, вакцинация человека и животных и пассивная иммунизация) не оказывают терапевтического эффекта.
Болезнь Крейтцфельдта — Якоба считается летальным заболеванием. Продолжительность жизни большинства пациентов в основном не превышает 1 года с момента манифестации клинических проявлений, средняя продолжительность жизни составляет 8 месяцев. И лишь 5−10% больных продолжают жить в течение 2 и более лет.
Болезнь Крейтцфельдта-Якоба
Болезнь Крейтцфельдта-Якоба — редко встречающееся дегенеративное заболевание головного мозга, связанное с накоплением в нейронах патологического белка приона. Клинически болезнь Крейтцфельдта-Якоба проявляется слабоумием, пирамидными и экстрапирамидными нарушениями, миоклониями, симптомами поражения мозжечка и нарушением зрения. Диагноз болезни Крейтцфельдта-Якоба основывается на совокупности клинических симптомов, данных ЭЭГ, анализа цереброспинальной жидкости, МРТ и ПЭТ головного мозга, а также морфологического исследования образца тканей мозга, полученного в результате биопсии или посмертно. Эффективное лечение болезни Крейтцфельдта-Якоба пока не найдено. Заболевание имеет 100% летальный исход.
Болезнь Крейтцфельдта-Якоба очень редкое заболевание. Ранее в литературе указывалось, что частота его встречаемости примерно 1 случай на 1 млн. человек населения.
Однако в 90-х годах прошлого века начали отмечаться случаи так называемого нового варианта болезни Крейтцфельдта-Якоба, связанного с заражением от крупного рогатого скота и названного «коровьим бешенством». Только в Англии за 5 лет от этого заболевания умерло 86 человек.
Наиболее распространена болезнь Крейтцфельдта-Якоба среди людей в возрасте 65-70 лет и старше. Однако в новом варианте болезнь Крейтцфельдта-Якоба зачастую поражает лиц молодого возраста. Относительно высокий уровень заболеваемости отмечается в Англии, Израиле, Чили и Словакии.
Болезнь Крейтцфельдта-Якоба
Установлено, что болезнь Крейтцфельдта-Якоба имеет инфекционный характер.
Заражение может произойти при пересадке зараженных прионами тканей, через нейрохирургический инструмент и препараты крови, при введении некоторых гормональных препаратов (человеческого гонадотропина для лечения бесплодия и соматотропина для терапии гипопитуитаризма). Болезнь Крейтцфельдта-Якоба новой формы может развиваться после употребления в пищу мяса заболевших животных (коровы) или носителей инфекции (овец и коз).
В результате ряда исследований стало известно, что болезнь Крейтцфельдта-Якоба связана с проникновением в организм инфекционного белка — приона. В норме в клетках головного мозга человека содержится здоровый прион, имеющий несколько другое строение.
Инфекционный прион, попадая в организм человека не разрушается, а с током крови поступает в головной мозг и откладывается на поверхности нейронов.
Его взаимодействие с нормальными прионами мозговой клетки приводит к тому, что они изменяют свою структуру, постепенно трансформируясь в патогенную, подобную инфекционному приону, форму. Патогенные прионы образуют бляшки и приводят к гибели нейрона.
Болезнь Крейтцфельдта-Якоба имеет достаточно длительный инкубационный период, связанный с временем, необходимым для проникновения инфекционных прионов в мозговую ткань и патогенной трансформации здоровых прионов.
Длительность инкубационного периода напрямую зависит от способа заражения. При инфицировании тканей головного мозга зараженным хирургическим инструментом болезнь Крейтцфельдта-Якоба развивается через 15-20 месяцев.
При инфицировании через имплантированные в околомозговые структуры ткани (например, твердую мозговую оболочку, роговицу глаза) инкубационный период может длиться до 5,5 лет.
При внутримышечном введении инфицированных лекарственных препаратов (например, гонадотропина, соматотропина, содержащих бычий тромбин гемостатиков) болезнь Крейтцфельдта-Якоба начинает проявляться спустя 12,5 лет.
Отмечаются также наследственные формы болезни Крейтцфельдта-Якоба, связанные с генетическими нарушениями, приводящими к образованию патологических прионов.
Практическая неврология классифицирует болезнь Крейтцфельдта-Якоба с учетом ее клинической формы.
В соответствии с этим выделяют: подострую спонгиоформную энцефалопатию, отличающуюся быстрым течением и диффузным поражением мозговой коры; классическую (дискинетическую) форму, представляющую собой сочетание пирамидных и экстрапирамидных симптомов со слабоумием (деменцией); промежуточную форму болезни Крейтцфельдта — Якоба, характеризующуюся преобладанием мозжечковых и подкорковых расстройств; амиотрофическую форму, двигательные и речевые расстройства при которой напоминают клинику бокового амиотрофического склероза.
Различают также наблюдавшуюся ранее спорадическую форму заболевания и новый вариант болезни Крейтцфельдта-Якоба («коровье бешенство»).
В большинстве случаев болезнь Крейтцфельдта-Якоба характеризуется постепенным развитием, однако возможно подострое или острое начало.
Примерно в 30% случаев болезнь Крейтцфельдта-Якоба начинается с продромальных симптомов: раздражительности, рассеянности, головных болей, нарушений сна, головокружения, ухудшения памяти, снижения зрения, безынициативности, снижения либидо, изменения поведенческих реакций.
Возможно эпизодическое возникновение эйфории и/или беспричинного страха, отрывистые бредовые или галлюцинаторные переживания.
Из неврологических нарушений в продромальном периоде наблюдаются: шаткость во время ходьбы, парестезии, расстройство высших функций коры головного мозга (алексия, акалькулия и пр.). Описаны несколько случаев, когда болезнь Крейтцфельдта-Якоба дебютировала с появления корковой слепоты.
В стадии развернутых клинических проявлений болезнь Крейтцфельдта-Якоба характеризуется прогрессирующим спастическим параличем (параплегией или гемиплегией), атаксией, эпилептическими припадками. Возникают экстрапирамидные нарушения: мышечная ригидность, атетоз, тремор.
Практически у всех больных наблюдаются миоклонии — быстрые неритмичные сокращения отдельных мышц. Чаще всего отмечается миоклонус губы и века. Наблюдаются вторично генерализованные миоклонические приступы.
Появляется и нарастает ярко выраженная деменция, сопровождающаяся нарушениями речи вплоть до ее полного распада. Новый вариант болезни Крейтцфельдта-Якоба отличается преобладанием психиатрической симптоматики и расстройств чувствительности.
В 100% случаев он сопровождается мозжечковыми нарушениями, в то время как при спорадической болезни Крейтцфельдта-Якоба расстройства функции мозжечка наблюдаются лишь в 40% случаев.
В терминальной стадии болезнь Крейтцфельдта-Якоба характеризуется глубокой деменцией. Пациенты не контактны, находятся в состоянии прострации, утрачен контроль над функцией тазовых органов.
Наблюдаются гиперкинезы, выраженные мышечные атрофии, нарушения глотания, пролежни. Возможна гипертермия и эпилептические приступы.
Смерть наступает в коматозном состоянии на фоне децеребрационной ригидности и выраженной кахексии.
Клиническая диагностика заболевания основана на сочетании прогрессирующей в течение 2-х лет деменции, пирамидных и экстрапирамидных расстройств, миоклоний, мозжечковых расстройств и нарушений зрения.
Для уточнения диагноза невролог назначает инструментальные методы обследования: электроэнцефалографию (ЭЭГ), ПЭТ и МРТ головного мозга, люмбальную пункцию.
В сомнительных случаях для установления диагноза болезнь Крейтцфельдта-Якоба производят стереотаксическую биопсию головного мозга.
На ЭЭГ на фоне сниженной биоэлектрической активности у большинства больных наблюдаются периодические или псевдопериодические острые волны.
Отмечается билатеральная, фокальная или генерализованная миоклоническая активность, которая в начальной стадии определяется у половины больных, а в терминальной стадии выявляется в 100% случаев спорадической болезни Крейтцфельдта-Якоба. Новый вариант заболевания часто протекает без существенных изменений ЭЭГ-паттерна.
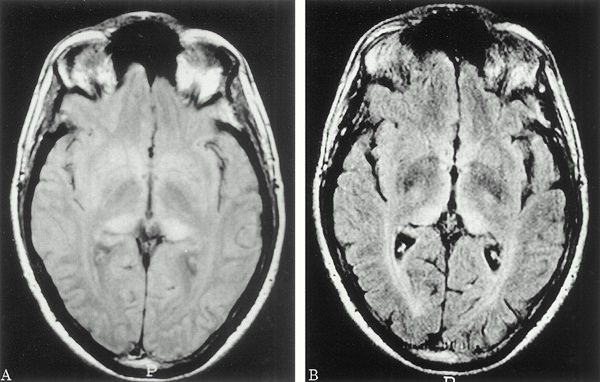
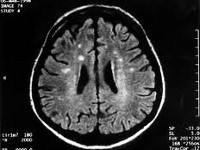
При проведении МРТ головного мозга в Т2-режиме определяется так называемый «симптом медовых сот» – участки повышенного сигнала, исходящие от подкорковых ганглиев и таламуса.
Зачастую выявляются признаки атрофических изменений мозжечка и коры головного мозга, расширение желудочков и боковых цистерн мозга.
ПЭТ диагностирует зоны пониженного метаболизма, локализующиеся в подкорковых ядрах, полушариях мозжечка и коре мозга.
Люмбальная пункция в обязательном порядке проводится пациентам с подозрением на болезнь Крейтцфельдта-Якоба. Она позволяет оценить давление ликвора и произвести исследование цереброспинальной жидкости. Отсутствие патологических изменений ликвора позволяет дифференцировать болезнь Крейтцфельдта-Якоба от многих других заболеваний ЦНС.
Наиболее достоверным методом диагностики является морфологическое исследование образцов мозговой ткани, которые могут быть получены прижизненно путем биопсии или при аутопсии после смерти пациента. Применение иммуноцитохимического метода позволяет обнаружить в исследуемом материале отложения патологического белка — приона.
Дифференциальную диагностику болезни Крейтцфельдта-Якоба необходимо проводить с лобно-височной деменцией, герпевирусным энцефалитом, болезнью Альцгеймера, мультиинфарктной деменцией (слабоумием, развивающимся после повторных ишемических и геморрагических инсультов), хроническим менингитом, арахноидитом, нормотензивной гидроцефалией, энцефалопатией Хашимото, сопровождающей некоторые случаи аутоиммунного тиреоидита, и др.
В современной медицине подходы к лечению болезни Крейтцфельдта-Якоба находятся в стадии активной разработки. Общепринятые противовирусные препараты, а также пассивная иммунизация и вакцинация людей и животных выявились неэффективными.
Отмечено, что блокирующее действие на синтез патологических прионов в инфицированных нейронах оказывает Брефелдин А, а блокаторы кальциевых каналов продлевают жизнь инфицированных клеток. Обычно пациенты, имеющие болезнь Крейтцфельдта-Якоба, получают симптоматическое лечение.
Оно направлено на купирование миоклонических приступов и экстрапирамидных нарушений, в связи с чем применяются антиэпилептические и противопаркинсонические лекарственные средства.
Болезнь Крейтцфельдта-Якоба является фатальным заболеванием. Продолжительность жизни большинства больных не превышает 1 год с момента начала клинических проявлений; а средняя длительность составляет 8 месяцев. Лишь 5-10% заболевших живут в течение 2 и более лет. Наследственная болезнь Крейтцфельдта-Якоба в среднем длится около 26 месяцев.
Болезнь Крейтцфельдта-Якоба
Болезнь Крейтцфельдта-Якоба — это тяжелое дегенеративное заболевание постоянно прогрессирующего характера, локализуется в базальных ганглиях, спинном или головном мозге. Другие названия патологии: псевдосклероз спастический, трансмиссивная спонгиоформная энцефалопатия, синдром кортико-стриоспинальной дегенерации, коровье бешенство.
статьи:
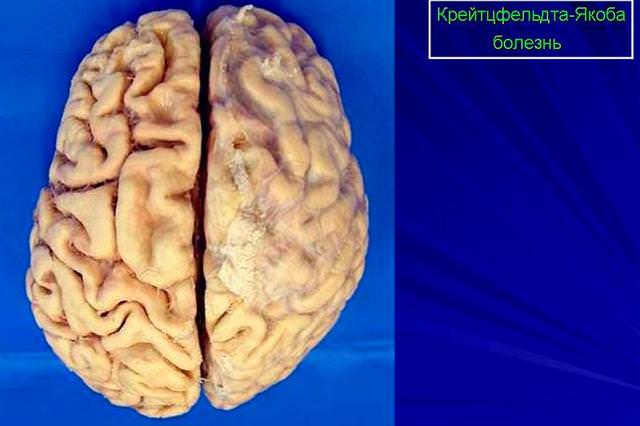
Данное заболевание составляет 85% всех прионовых энцефалопатий, встречающихся у людей. Оно поражает одинаково интенсивно представителей всех рас, полов и возрастных групп. Незначительное преобладание частоты заболевания отмечается среди женщин на острове Новая Гвинея, с чем современные ученые часто связывают случаи каннибализма, традиционные для населения указанных территорий.
Попадая в человеческий организм, прион, который представляет собой инфекционную субмикроскопическую частицу, вызывающую дегенерацию головного мозга, задерживается на поверхности клетки и взаимодействует со здоровыми белками.
В результате их структура нарушается настолько, что внешне мозг человека напоминает губку. При этом естественные процессы в его больных клетках блокируются, что вызывает их быстрое разрушение.
Здоровые ткани, находящиеся рядом с очагом заболевания, воспаляются, и этот процесс приводит к постоянному усугублению состояния пациента.
Причины болезни Крейтцфельдта-Якоба
Основной причиной данного расстройства считается поедание мяса животных, болеющих спонгиозной энцефалопатией.
Кроме того, были отмечены случаи заражения пациентов в медицинских учреждениях через некачественно стерилизованные хирургические инструменты или через трансплантируемые ткани.
Описаны эпизоды заражения людей, подвергавшихся гормонотерапии, направленной на устранение дефицита гормона роста, когда лечение производилось с помощью экстрактов тканей, содержащих в себе данный гормон.
Кроме случаев инфицирования извне, существует наследственный фактор, вызывающий болезнь Крейтцфельдта—Якоба. Данная форма заболевания связана с мутацией в гене PRNP. Это нарушение вызывает появление в организме патологических прионов. Наследственное заболевание наблюдается в 10-15% семей, в которых когда-либо был поставлен диагноз любой формы данного расстройства.
Классификация болезни Крейтцфельдта—Якоба
Согласно классификации, заболевание Крейтцфельдта—Якоба подразделяется на 4 типа:
Симптомы болезни Крейтцфельдта–Якоба
Для раннего периода заболевания характерен такой симптом, как изменение личности, при котором человек проявляет беспокойство. Также для этой стадии характерны депрессии, бессонница, забывчивость и значительное ухудшение интеллектуальных способностей. Постепенно больной теряет зрение, что способствует утрате возможности адекватно обслуживать себя.
За этим следуют затруднения проглатывания пищи и воды, появления непроизвольных резких движений, которые невозможно предугадать. С прогрессированием заболевания состояние пациента ухудшается, что влечет за собой его впадение в кому.
Из этого состояния человек уже не выходит, а смерть наступает в результате развивающейся сердечной недостаточности, пневмонии и присоединения других инфекций.
У всех пациентов психиатрические симптомы присутствуют в начале заболевания, а позднее наступает фаза деменции. Поскольку болезнь неизлечима ее исход всегда только летальный.
Диагностика болезни Крейтцфельдта–Якоба
Способов постановки точного диагноза данной болезни в начале ее течения на сегодняшний день не существует. Специалисты могут предположить заболевание с помощью семейного анамнеза, неврологических тестов и некоторых диагностических процедур.
Лечение заболевания Крейтцфельдта – Якоба
В современной медицине не существует способов успешного излечения пациентов от данного заболевания. В последние десятилетия учеными были проведены тесты множества препаратов: антибиотиков, стероидов, иммуномодуляторов и антивирусных лекарств. Но все рассматриваемые методы показали полное отсутствие терапевтического эффекта.
Сегодня, работая с пациентами, страдающими болезнью Крейтцфельдта—Якоба, врачи могут проводить исключительно симптоматическое лечение, направленное на облегчение тяжести течения заболевания, но не на уничтожение прионов и оздоровление мозга. При подозрении на КБЯ, больному отменяют лекарственные препараты, негативно воздействующие на мнестические функции его организма.
Положительное влияние на состояние пациентов, а именно облегчение симптомов заболевания было выявлено при использовании следующих медикаментов: Брефелдина А и блокаторов кальциевых каналов.
Первые, разрушая аппарат Гольджи, создают препятствие для выработки PrPSc в структуре зараженных клеток.
Вторые останавливают работу NMDA-рецепторов, что становится причиной более длительного срока жизни инфицированных нейрональных культур.
Прогноз болезни Крейтцфельдта—Якоба
Практически во всех случаях данное заболевание приводит к смерти пациента в течение одного года.
Иногда летальных исход наступает ранее — через полгода после того, как был поставлен диагноз, но в особо редких случаях — приблизительно в 5% случаев — продолжительность заболевания может увеличиться до двух лет.
10% заболевших людей не проживают и нескольких недель, что вызвано индивидуальными особенностями их организмов.
Поскольку болезнь не передается воздушно-капельным путем, для медицинских работников и родственников, контактирующих с больными, ношение масок и других защитных средств не является обязательным условием.
Те сотрудники, которые проводят анализы биологических жидкостей и тканей пациентов, у которых подозревается болезнь Крейтцфельдта—Якоба, должны соблюдать правила безопасности и работать исключительно в перчатках. В особенности необходимо избегать прямого контакта слизистых оболочек с образцами зараженного материала.
Если такой крайне нежелательный контакт все же состоялся, необходимо в обязательном порядке произвести дезинфекцию 4% раствором гидроксида. Обработка проводится в течение 5-10 минут после контакта, после чего следует промывание слизистой проточной водой.
Обеззараживание инструментов проводится автоклавированием в течение часа при температуре 132°С либо часовой стерилизацией в 10% растворе гипохлорита натрия или 4% растворе гидроксида натрия. Стандартные способы стерилизации инструментов, такие как обработка формалином, в данном случае неэффективны.
Болезнь Крейтцфельдта-Якоба: причины, симптомы, прогноз жизни
Болезнь Крейтцфельдта-Якоба была описана в начале 20 века двумя немецкими неврологами A. Jacob и H. Creutzfeldt.
Считается самым частым прионным заболеванием, среди встречающихся у человека (частота достигает одного случая на 1000000 человек населения).
- Причины
- Симптомы
- Диагностика
- Прогноз жизни
- Лечение
- Факты
Информация для врачей. При шифровке диагноза используется код A81.0. Указывается спорадический (идиопатический), семейный или ятрогенный случай. В последнем варианте указывается источник инфекции. Также, у пациентов со спорадической формой по возможности указывают тип заболевания.
По мнению большинства исследователей, считается следствием генной мутации, которая развивается под воздействием прионов (мелких белковых структур, в определенных условиях вызывающих хронические инфекционные заболевания). Обычны спорадические случаи возникновения заболевания, однако имеют место и семейные формы.
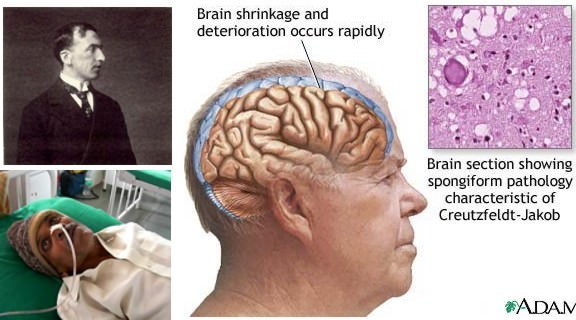
Причины
Главной причиной заболевания считается употребление в пищу мяса болеющих спонгиозной энцефалопатией животных. Также описаны случаи передачи болезни через зараженные хирургические инструменты, при пересадке ткани (например, трансплантация роговицы от молодых быков). Также описаны единичные случаи развития заболевания при гормонотерапии экстрактами тканей, содержащих гормон роста.
Симптомы
Заболевание медленно, но неуклонно прогрессирует. Острое начало встречается крайне редко. У 30-40% отмечается период продромы с наличием астенического, депрессивного синдрома, снижения аппетита, расстройства внимания. Такой период может продолжаться до нескольких месяцев.
Дебют болезни Крейтцфельдта-Якоба выражается в развитии интеллектуальных, аффективных нарушений. Также больного начинают беспокоить головные боли, головокружение, нарушения речи, нарушения зрения. В неврологическом осмотре можно определить наличие атаксии, изменения полей зрения. Иногда у пациента имеют место миоклонии.
Со временем прогрессирования заболевания развивается центральный парез конечностей, развиваются выраженные экстрапирамидные нарушения, могут развиваться генерализованные судорожные припадки, нарастает деменция по альцгеймеровскому типу. В конечном итоге развивается кома и позже наступает смерть.
Клинически можно выделить шесть основных форм болезни Крейтцфельдта-Якоба, в зависимости от выраженности тех или иных симптомов:
- Окципитальная (по автору – Хейденхайна): поражаются задние отделы коры, рано развивается слепота.
- Атаксическая (Броунелла-Оппенгеймера): поражается ствол, мозжечок, главный симптом – атаксия.
- Экстрапирамидная (Стерна-Гарсиа): поражаются базальные ганглии, на первый план выходят явления паркинсонизма.
- Фронтальная (Якоба): поражается лобная доля, превалируют психические нарушения.
- Амиотрофическая: встречается редко, поражаются нейроны передних рогов (развитие двигательных расстройств).
- Панэнцефалопатическая (Мизутани): диффузное поражение серого и белого вещества с разнообразной клинической картиной, выраженными общемозговыми нарушениями.
Диагностика
Диагностика болезни Крейтцфельдта-Якоба основывается на данных клинической картины, неврологического статуса, случаев употребления мяса животных из очагов эпидемий спонгиозной энцефалопатии животных.
Также необходимо исключение другой патологии, которая могла бы привести к клиническим проявлениям заболевания. Так, при ЭЭГ исследовании выявляются фоновые плоские колебания в виде волн, состоящих из трех фаз. На эти волны могут накладываться острые и медленные высокоамплитудные волны с частотой около 2 Гц.
МСКТ и МРТ исследование выявляет признаки дистрофии ткани мозга. Кровь и ликвор, как правило, не меняются.
Достоверным диагноз считается после проведения прижизненной биопсии мозга с использованием специальных молекулярных методов исследования.
В случае невозможности проведения исследования, несогласии больного или его законных представителей, диагноз считается вероятным, но не подтвержденным.
Прогноз жизни
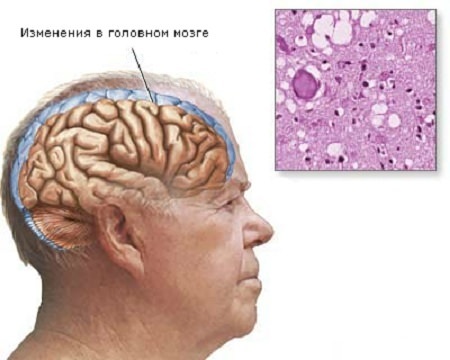
Конечная стадия болезни Крейтцфельдта-Якоба представляет собой глубокую деменцию, коматозное состояние. В мозговом веществе развивается губчатая (спонгиозная) дегенерация. Также развивается глиоз, нередка вакуолизация структур мозга. Продолжительность заболевания редко превышает 1 год.
Лечение
Специфической терапии не существует. Болезнь Крейтцфельдта-Якоба в 100% случаев приводит к летальному исходу.
Необходима симптоматическая терапия, поддержание жизненных функций в коматозном состоянии. По некоторым данным имеет некоторый эффект препарат Брефелдин А.
Также высказываются предположения об увеличении продолжительности жизни нейрональных структур при приеме блокаторов кальциевых каналов.
Факты
В 1896 году была описана вспышка заболевания спонгиозной энцефалопатии коров в Великобритании. В 1990 же годах описано заболевание, схожее по клинике с болезнью Крейтцфельда-Якоба у 21 человека, из них 20 умерло к 1997 году. Однако на ЭЭГ не было характерных для болезни изменений.
Алексей Борисов (врач-невролог)
Практикующий врач-невролог. Окончил Иркутский государственный медицинский университет. Работает в факультетской клинике нервных болезней. Подробнее…
Болезнь Крейтцфельдта-Якоба – Все о массаже..
Болезнь Крейтцфельдта-Якоба — данное заболевание носит тяжелый дегенеративный характер, при этом она постоянно прогрессирует.
Локализуется в базальных ганглиях, спинном или головном мозге.
У этого заболевания есть также и другие названия: псевдосклероз спастический, трансмиссивная спонгиоформная энцефалопатия, синдром кортико-стриоспинальной дегенерации, коровье бешенство.
Болезнь Крейтцфельдта-Якоба (БКЯ) довольно-таки редко встречающееся дегенеративное заболевание головного мозга, связанное с накоплением в нейронах патологического белка приона. Клиническое проявление болезни Крейтцфельдта-Якоба обозначается слабоумием, пирамидными и экстрапирамидными нарушениями, миоклониями, симптомами поражения мозжечка и нарушением зрения.
Первое описание болезни, было сделано еще в 1920 г. немецким невропатологом Гансом Герхардом Крейтцфельдтом, а изучавший это же заболевание немецкий невропатолог Альфонс Якоб в 1921 г.
обнаружил сочетание возникающих при этой патологии нарушений психики с симптомами, возникающими при поражении пирамидной и экстрапирамидной систем, а также передних рогов спинного мозга. В своем описании А.
Якоба, данное заболевание определяет как псевдосклероз спастический. Выделяют несколько стадий заболевания.
Классификация болезни Крейтцфельдта—Якоба
Согласно классификации, заболевание Крейтцфельдта—Якоба подразделяется на 4 типа:
Классический.
В данной форме заболевания прионы поражают головной мозг без видимых на то причин. Подобный вид коровьего бешенства имеет место быть, как правило у людей старше 50 лет, и встречается крайне редко — в 1-2 случаях на миллион.
Проявляется инфекция полной или частичной потерей памяти, внезапными переменами настроения, снижением интереса к жизни. Впоследствии больной утрачивает способность обслуживать себя, теряет зрение и практически перестает говорить.
На последних стадиях пациента беспокоят сильные галлюцинации.
- Наследственный.
- Течение стадий заболевания аналогично классическому типу, но причиной, вызвавшей болезнь, является генетическое наследование прионов.
- Ятрогенный.
Данным типом заболевания пациент заражается при оперативном вмешательстве через лекарства, плохо стерилизованные инструменты, оболочки мозга, применяемые для закрытия ран. В 21 веке вероятность заражения болезнью Крейтцфельдта—Якоба во время операции практически нулевая.
Новый вариант.
Форма заболевания, при которой заражение происходит через мясную пищу, содержащую зараженные ткани. Известно о более чем ста случаях заболевания с 1995 года. Болезнь особенно часто может поражать молодых людей, средний возраст которых равен 20 годам.
Начальная стадия заболевания отмечается депрессией, потерей интереса к увлечениям и работе, замкнутостью и желанием одиночества. Далее наступает нарушение координации движений, отсутствие возможности самостоятельно есть и приводить себя в порядок.
В отличие от классического типа, при данном варианте заболевания слабоумие наступает в последнюю очередь, и пациент осознает все происходящее с ним.
Также установлено, что болезнь Крейтцфельдта-Якоба имеет инфекционный характер.
Заражение может произойти при пересадке зараженных прионами тканей через нейрохирургический инструмент и препараты крови, при введении некоторых гормональных препаратов (человеческого гонадотропина для лечения бесплодия и соматотропина для терапии гипопитуитаризма). Болезнь Крейтцфельдта-Якоба новой формы может развиться после употребления в пищу мяса заболевших животных (коровы) или носителей инфекции (овец и коз).
В результате проведения ряда исследований стало известно, что болезнь Крейтцфельдта-Якоба связана с проникновением в организм инфекционного белка — приона. В своей норме в клетках головного мозга человека содержится здоровый прион, который имеет несколько другое строение.
Инфекционный прион при попадании в организм человека не разрушается, а с током крови поступает в головной мозг и там откладывается на поверхности нейронов. Его взаимодействие с нормальными прионами мозговой клетки приводит к тому, что они изменяют свою структуру, постепенно трансформируясь в патогенную, подобную инфекционному приону форму.
Патогенные прионы вскоре образуют бляшки и приводят к гибели нейрона.
Болезнь Крейтцфельдта-Якоба имеет достаточно продолжительный инкубационный период. Это связанно со временем, которое необходимо для проникновения инфекционных прионов в мозговую ткань и патогенной трансформации здоровых прионов. Продолжительность инкубационного периода напрямую зависит от того, каким способом было получено заражение.
В случае инфицировании тканей головного мозга, зараженным хирургическим инструментом, болезнь Крейтцфельдта-Якоба развивается через 15-20 месяцев.
А если инфицирование произошло через имплантированные в околомозговые структуры ткани (например твердую мозговую оболочку, роговицу глаза), то тогда инкубационный период может длиться до 5,5 лет.
При внутримышечном введении инфицированных лекарственных препаратов (например гонадотропина, соматотропина, содержащих бычий тромбин гемостатиков) болезнь Крейтцфельдта-Якоба может начать свое проявление спустя 12,5 лет.
Отмечаются также наследственные формы болезни Крейтцфельдта-Якоба, связанные с генетическими нарушениями, которые приводят к образованию патологических прионов.
Симптомы болезни Крейтцфельдта-Якоба
В большинстве случаев болезнь Крейтцфельдта-Якоба может охарактеризовываться постепенным развитием, но также возможно подострое или острое начало.
Примерно в 30% случаев болезнь Крейтцфельдта-Якоба может начинаться с продромальных симптомов – это раздражительность, рассеянность, головные боли, нарушение сна, головокружение, ухудшение памяти, снижение зрения, безынициативность, снижение либидо, изменение поведенческих реакций.
Возможно также проявление эпизодического возникновения эйфории или беспочвенного страха, отрывистые бредовые или галлюцинаторные переживания.
Из неврологических нарушений в продромальном периоде, могут также отмечаться: шаткость во время ходьбы, парестезии, расстройство высших функций коры головного мозга (алексия, акалькулия). Были даже описаны несколько случаев, когда болезнь Крейтцфельдта-Якоба дебютировала с появления корковой слепоты.
В стадии развернутых клинических проявлений болезнь Крейтцфельдта-Якоба характеризуется прогрессирующим спастическим параличем (параплегией или гемиплегией), атаксией, эпилептическими припадками.
Как правило возникают экстрапирамидные нарушения: мышечная ригидность, атетоз, тремор. Отмечено, что практически у всех больных наблюдаются миоклонии — быстрые неритмичные сокращения отдельных мышц.
Также весьма часто отмечается миоклонус губы и века.
Могут наблюдаться вторичные генерализованные миоклонические приступы. Проявляется и нарастает ярко выраженная деменция, которая сопровождается нарушениями речи, даже может привести к ее полному распаду.
Новый вариант болезни Крейтцфельдта-Якоба отличается преобладанием психиатрической симптоматики и расстройств чувствительности.
В 100% случаев этот вариант сопровождается мозжечковыми нарушениями, хотя при спорадической болезни Крейтцфельдта-Якоба расстройства функции мозжечка наблюдаются лишь в 40% случаев.
В терминальной стадии болезнь Крейтцфельдта-Якоба характеризуется глубокой деменцией. Пациенты становятся не контактны, находятся в состоянии прострации, теряется контроль над функцией тазовых органов.
Отмечаются гиперкинезы, ярко выраженные мышечные атрофии, нарушения глотания, пролежни. Также возможна гипертермия и эпилептические приступы.
Смерть у таких пациентов наступает в коматозном состоянии на фоне децеребрационной ригидности и выраженной кахексии.
Диагностика болезни Крейтцфельдта–Якоба
Способов постановки точного диагноза данной болезни в начале ее течения, к сожалению на сегодняшний день не существует. Специалисты могут только предположить заболевание с помощью семейного анамнеза, неврологических тестов и некоторых других диагностических процедур.
Конечно в первую очередь больной будет направлен на электроэнцефалографию. Во время данной процедуры на голову обследуемого больного прикрепляются электроды, которые служат для измерения электрической активности головного мозга. В случае наблюдения нарушений в данной активности могут указывать на то, что спровоцированы они болезнью Крейтцфельдта—Якоба.
Помимо электроэнцефалографии о развитии данного заболевания могут также свидетельствовать и результаты МРТ.
Такое исследование основывается на применение магнитных полей высокой мощности для создания изображения тканей мозга на мониторе.
Применяя этот способ, можно получить предельно качественную картинку серого и белого вещества, при следующем за тем осмотре можно выявить изменения, которые являются признаками заболевания.
Для постановки возможного диагноза проводится также анализ спинномозговой жидкости. Для этого производится люмбальная пункция и исследование полученного ее путем материала. Здесь признаком заболевания может быть наличие протеина, которого в здоровом состоянии быть не должно.
Также применяется еще один этап обследования — биопсия миндалин, ткани которых способны накапливать в себе признаки прионной инфекции. Правда этот метод диагностики заболевания считается самым ненадежным из всех перечисленных.
Наиболее надежным анализом при диагностировании коровьего бешенства врачи предпочитают считать биопсию мозга. Данная процедура основывается на том, что у пациента забирают небольшое количество клеток с помощью специальной иглы. Данный метод дает возможность осмотреть пораженную ткань вблизи и сделать самые точные выводы о том, насколько и какой инфекцией поражен головной мозг.
Лечение болезни Крейтцфельдта–Якоба
Как правило, при данном недуге терапии нет. Проводится только симптоматическое лечение. В случае выявлении БКЯ необходимо отменить все лекарственные препараты, ведь их действие может негативно влиять на мнестические функции и поведение пациента.
Разнообразное множество потенциальных терапевтических вмешательств при БКЯ остаются в настоящее время на уровне дискуссии.
Традиционные же противовирусные средства, такие как амантадин, интерфероны, пассивная иммунизация и вакцинация человека и животных, оказались вообще неэффективными.
Профилактика болезни Крейтцфельдта–Якоба
Так как коровье бешенство у людей развивается после употребления мяса инфицированного скота, то введен запрет на экспорт мясопродуктов из тех регионов, где были зафиксированы случаи этого заболевания. Категорически запрещено также использование костной муки при производстве животных кормов и продажа мяса животных, чей возраст превышает 2 года.
Те, кто имеют контакт с биологическим материалом, а именно медработники, должны пользоваться перчатками, а в случае попадания зараженных жидкостей или тканей на кожу, проводить 5-10 мин.
дезинфекцию 4 % раствором каустической соды и промывать это место проточной водой.
Можно также для дезинфекции использовать применяющийся для дезинфекции на предприятиях мясной промышленности анолит, сочетающий стерилизующие и моющие свойства и эффективно уничтожающий бактериальных и вирусных возбудителей.
Материалы и инструменты настоятельно рекомендуется дезинфицировать раствором гидроксида натрия на протяжении часа. Разрушается третичная структура прионов также воздействиями сильнокислых моющих веществ и при обработке хлорной известью.
Такое заболевание, как болезнь Крейтцфельдта-Якоба является фатальным заболеванием. Продолжительность жизни большинства больных не превышает 1 год с момента начала клинических проявлений; а средняя длительность составляет 8 месяцев. Лишь 5-10% заболевших живут в течение 2 и более лет.
Наследственная болезнь Крейтцфельдта-Якоба в среднем длится около 26 месяцев.
Нужно внимательней быть всем нам к таким опасным недугам. А уж тем людям, которые непосредственно могут быть в контакте с таким заболеванием тем более.
Comments
(0 Comments)