Болезнь баттена, нцл 2 типа: симптомы и лечение
Болезнь Баттена (нейронный цероидный или восковидный липофусциноз) – нейродегенеративное наследственное заболевание, которое встречается очень редко и проявляется в виде постоянного наращивания липопигментов в нервных тканях. Этот недуг относится к группе липофусцинозов – лизосомных болезней при отклонениях метаболизма из-за поломок генов, во время которых липидные молекулы накапливаются внутри клеток тканей, постепенно их разрушая.

Классификация заболевания насчитывает четыре разновидности:
- Болезнь Сантавуори-Халтиа (НЦЛ 1 типа) с высокой скоростью развития, проявляющаяся у детей младенческого возраста от шести до двадцати четырёх месяцев с продолжительностью жизни чуть больше пяти лет, которая имеет следующую симптоматику: микроцефалия (у ребёнка маленькая голова вследствие недоразвитости головного мозга с отсутствием или сглаживанием извилин) и миоклонические судороги.
- Поздняя инфантильная форма болезни Баттена или НЦЛ 2 типа (нейронного цероидного липофусциноза второго типа) развивается только к двум – четырём годам со следующими симптомами: судороги, которые никоим образом не реагируют на приём противосудорожных лекарств, и атаксия — потеря координации.
- Ювенальная форма НВЛ (НЦЛ 3), именуемая также синдромом Шпильмейера-Фогта-Шегрена, развивается ещё позже в пяти – десятилетнем возрасте с потерей зрения, судорогами, двигательными нарушениями.
- Взрослая форма НВЛ или болезнь Куфса проявляется до сорокалетнего возраста и имеет медленно прогрессирующую неврологическую симптоматику, связанную с гибелью нейронов головного мозга.
Иногда болезнь наблюдается с нетипичным течением, не подпадающая под вышеописанную классификацию с более быстрым или медленным течением, а также с изменением симптомов.
Нетипичная болезнь Баттена встречается у одного из десяти больных.
Причины возникновения и патогенез
Причина возникновения недуга – наследственное нарушение строения ДНК, которое передаётся по мужской и женской линиям. При этом у больного ребёнка нет гена NCL, отвечающего за выработку вещества, которое необходимо для переработки токсического липофусцина, который является жёлтым пигментом и вырабатывается при разложении отработавших митохондрий.

Липофусцин также называют пигментом старения или изнашивания. Повышенное содержание данного вещества наблюдается в старости или при заболеваниях с деградацией тканей.
В идеале, оно должно быть переработано митохондриями при помощи специального фермента TPP1, но так как гена, отвечающего за его продуцирование нет, то липофусцин накапливается как токсинный мусор в различных клетках и разрушает их (при болезни Баттена процесс накопления происходит в нейронах).
Из-за массовой гибели нейронов происходит атрофия мозжечка, а также больших полушарий, что хорошо различимо во время проведения магнитно-резонансной томографии.
Так как заболевание наследственное, то практически всегда проявляется в детском возрасте, при этом конкретный возраст появления первых симптомов зависит от скорости выработки липофусцина, поэтому, чем медленнее она происходит, тем позже проявляется заболевание и тем дольше жизнь больного с момента диагностики недуга (не считая атипичных случаев).
Симптомы
До накопления липофусцина в нейронах головного мозга больные дети не отличаются от своих сверстников.
По достижению количества этого вещества определённого порога, когда начинается гибель нейронов головного мозга начинают наблюдаться первые симптомы в виде отставания в развитии, эпилептических приступов, а также атаксии.
Атаксия – нарушение координаций движений с проявлениями мышечной слабости, вызванное поражением мозжечка, коры мозга или вестибулярного аппарата.
С прогрессией атрофии мозга начинаются умственные отклонения, потеря навыков, потеря зрения.
В конечном итоге заболевание приводит к полной потере мозговых функций сначала в виде обездвиженности, затем в отсутствии элементарных рефлексов и функций внутренних органов. Конечной стадией заболевания является кома, после которой наступает смерть из-за гибели головного мозга.
Так как гибель нейронов происходит неравномерно и у каждого ребёнка индивидуально, то качество проявлений симптомов, их последовательность и промежутки между стадиями могут быть у каждого разными.
Диагностика заболевания
Болезнь Баттена диагностируется прежде всего при помощи анализа ДНК, а также некоторых методов обследования:
- Общие анализы (вакуолизированные лимфоциты в крови).
- Осмотр у окулиста (дегенерация сетчатки).
- ЭЭГ (нарушение его нормальной электрической активности).
- МРТ мозга (его дегенеративные изменения).
- Биопсия тканей кожи и других (наличие в клетках сходного вещества с липофусином).
Лечение
На сегодняшний день это неизлечимое заболевание, так как пока не найдено абсолютно каких методов, способных хоть как-то остановить или замедлить процесс деградации нейронов. Единственное, что можно сделать максимально облегчить и продлить последние дни больного при помощи симптоматической и интенсивной терапии.
Прогноз
Прогноз при данном недуге зависит от каждого индивидуального случая, но обычно продолжительность жизни варьируется в зависимости от формы заболевания:
- При детской форме, диагностируемой до двухлетнего возраста, обычно умирают очень быстро, максимум до пяти лет.
- Поздняя инфантильная форма характеризуется немного большей длительностью – смерть наступает в 10-12 лет.
- С ювенальной формой доживают до двадцати лет, а в редких исключениях даже до тридцати.
- Пациенты со взрослой формой живут долго, так как болезнь прогрессирует медленно со слабым развитием симптоматики. Учитывая, что первые её проявления диагностируются во взрослом возрасте, больной вполне может дотянуть до старости.
- Продолжительность жизни у больных с атипичным проявлением болезни Баттена не определена и зависит от индивидуального случая.
Болезнь Баттена и восковидный липофусциноз нейронов
Что такое болезнь Баттена?
Болезнь Баттена – смертельное наследуемое заболевание нервной системы, которое обычно начинается в детстве.
Ранние симптомы этого заболевания как правило появляются в возрасте от 5 до 10 лет, когда родители или врачи начинают замечать, что у ранее нормального ребенка развиваются проблемы со зрением или судорожные приступы.
В некоторых случаях ранние симптомы достаточно неочевидные, которые могут проявляться в виде изменений личности и поведения, медлительности, проблем неуклюжести или спотыкания.
Со временем дети с болезнью Баттена сильнее страдают от когнитивных расстройств, происходит прогрессирование судорогу, ухудшается зрение и двигательные (моторные) навыки. В конце концов, дети с болезнью Баттена становятся слепыми, прикованными к постели и происходит потеря рассудка. Болезнь Баттена часто заканчивается смертельным исходом в позднем подростковом возрасте или в возрасте около двадцати лет.
Болезнь Баттена названа в честь британского педиатра, впервые описавшего ее в 1903 году.
Также известная как болезнь Шпильмейера-Фогта-Шёгрена-Баттена, это наиболее распространенная форма группы заболеваний, называемых восковидными липофусцинозами нейронов или НЦЛ.
Хотя болезнь Баттена первоначально относилась конкретно к несовершеннолетней форме NCL (JNCL), термин болезнь Баттена все чаще используется педиатрами для описания всех форм НЦЛ.
Другие формы восковидных липофусцинозов нейронов (НЦЛ, NCL)?
Существуют четыре других основных типа нейрональных липофусцинозов (НЦЛ), включая три формы, которые начинаются рано в детстве и очень редкая форма, поражающая взрослых. Симптомы данных детских форм НЦЛ подобны симптомам, вызванным болезнью Баттена, но они становятся явными в разном возрасте и прогрессируют с разной скоростью.
- Врожденная NCL – очень редкая и тяжелая форма восковидного липофусциноза нейронов. Младенцы имеют аномально маленькие головы (микроцефалию) и судороги, и умирают вскоре после рождения.
- Инфантильная форма нейронального липофусциноза (INCL или болезнь Сантавуори — Халтиа,) начинается между 6 и 2 годами и прогрессирует быстро. Страдающие заболеванием дети не могут развиваться и имеют микроцефалию. Также типичными являются короткие, острые мышечные сокращения, называемые миоклоническими судорогами или рывками. Дети с данной формой НЦЛ как правило умирают в возрасте до 5 лет, хотя некоторые из них прожили в вегетативном состоянии на несколько лет дольше.
- Поздняя инфантильная форма восковидного липофусциноза нейронов (LINCL, или болезнь Бильшовского-Янского) начинается между 2 и 4 годами. Типичными ранними признаками являются потеря координации мышц (атаксия) и судорог, которые не реагируют на медицинские препараты. Эта форма быстро развивается и заканчивается смертью в возрасте от 8 до 12 лет.
- Взрослая форма НЦЛ (также известная как болезнь Куфса, болезнь Парри и Adult NCL) обычно начинается до 40 лет, вызывает более мягкие симптомы, которые прогрессируют медленно и не вызывают слепоту. Хотя возраст смертей варьируется у пострадавших, эта форма сокращает продолжительность жизни.
- Существуют также «вариантные» формы поздней формы инфантильного восковидного липофусциноза нейронов (vLINCL), которые отличаются от классического позднего инфантильного НЦЛ.
Эпидемиология болезни Баттена и восковидного липофусциноза нейронов
Сколько людей имеют данные заболевания?
Болезнь Баттена и другие формы НЦЛ являются относительно редкими заболеваниями, встречаются, по статистике, от 2 до 4 из каждых 100 000 живорождений в Соединенных Штатах.
Данные заболевания, как представляется, более распространены в Финляндии, Швеции, других частях Северной Европы и Ньюфаундленде, Канада.
Хотя нейрональный липофусциноз классифицируются как редкие заболевания, они часто поражают более одного человека в семьях, которые несут дефектные гены.
Как наследуются восковидные липофусцинозы нейронов?
Детские формы НЦЛ – аутосомно-рецессивные расстройства, то есть они происходят только тогда, когда ребенок наследует две копии дефектного гена, по одному от каждого родителя. Когда оба родителя несут один дефектный ген, каждый из их детей сталкивается с одним из четырех шансов на развитие липофусциноза нейронов.
В то же время каждый ребенок также сталкивается с одним из двух шансов наследовать только одну копию дефектного гена. Люди, у которых есть только один дефектный ген, известны как носители, то есть они не развивают болезнь, но могут передавать ген своим собственным детям.
Поскольку мутированные гены, которые участвуют в определенных формах болезни Баттена, известны, в некоторых случаях возможно обнаружение носителей.
Взрослая форма НЦЛ может наследоваться как аутосомно-рецессивный или, реже, как аутосомно-доминантное расстройство. При аутосомно-доминантном наследовании у всех людей, которые наследуют одну копию гена болезни, развивается болезнь. В результате не существует незатронутых носителей этого гена.
Причины болезни Баттена и других форм восковидных липофусцинозов нейронов
Симптомы болезни Баттена и других НЦЛ связаны с накоплением веществ, называемых липофусцинами (липопигменты) в тканях организма. Эти липопигменты состоят из жиров и белков.
Их имя исходит из технического слова lipo, которое не подходит для «липидов» или жира, а также от термина «пигмент», используемого, поскольку они приобретают зеленовато-желтый цвет, если смотреть под микроскопом с ультрафиолетовым светом. Липопигменты накапливаются в клетках мозга и глаз, а также в коже, мышцах и многих других тканях.
Вещества находятся внутри части клеток, называемых лизосомами. Лизосомы выполняют функции очистки от элементов, которые становятся поврежденными или больше не требуются, и их необходимо очистить внутри клетки. Накопленные липопигменты в болезнь Баттена и другие восковидные липофусцинозы нейронов образуют отличительные формы, которые можно увидеть под электронным микроскопом.
Эти отложения – это то, что ищут врачи, когда они исследуют образец кожи для диагностики болезни Баттена. Конкретный вид липопигментных отложений может быть полезен для руководства дальнейшими диагностическими тестами, которые могут идентифицировать специфический дефект гена.
На сегодняшний день восемь генов связаны с различными формами восковидных липофусцинозов нейронов. Мутации других генов в NCL вероятны, поскольку некоторые люди не имеют мутаций ни в одном из известных генов. Более чем один ген может быть связан с конкретной формой НЦЛ. Известными генами восковидных липофусцинозов нейронов являются:
Диагностика
Поскольку потеря зрения часто является ранним симптомом, подозрение на болезнь Баттена может возникнуть во время обследований на глаза. Окулист может обнаружить потерю клеток в глазах, которая возникает при детских формах нейронального липофусциноза.
Однако, поскольку такая потеря клеток происходит также при других заболеваниях глаз, заболевание не может быть диагностировано только по этому признаку.
Специалист по глазным болезням либо другой врач, подозревающий НЦЛ, может направить ребенка к неврологу для дополнительного обследования.
Для диагностики восковидного липофусциноза нейронов, невропатологом проводится анализ медицинской и семейной истории пациента и информации из различных лабораторных исследований. Диагностические исследования, используемые для диагностики НЦЛ, включают в себя:
- Анализ крови или мочи. Данные анализы могут выявить аномалии, которые могут указывать на болезнь Баттена. Например, повышенные уровни химического вещества, называемого долихолом, обнаруживаются в моче многих людей с нейрональным липофусцинозом. Присутствие вакуолированных лимфоцитов – белых кровяных клеток, которые содержат дыры или полости (наблюдаемые микроскопическим анализом мазков крови) – при сочетании с другими результатами, которые указывают на NCL, направляет на вероятность наличия подростковой формы заболевания, вызванной мутациями CLN3.
- Отбор проб кожи или тканей. Врач может исследовать небольшой кусочек ткани под электронным микроскопом. Мощное увеличение микроскопа помогает врачу определить типичные осадки NCL. Эти отложения распространены в клетках кожи, особенно в потовых железах.
- Электроэнцефалограмма или ЭЭГ. ЭЭГ использует специальные электроды, размещенные на голове испытуемого, для записи электрических токов внутри мозга. Это позволяет врачам улавливать контрольные всплески электрической активности мозга, которые предполагают, что у человека возникают судороги.
- Электрические исследования глаз. Данные тесты, которые включают визуально реакции и электроретинограммы, могут обнаруживать различные проблемы с глазами, присущие детской форме восковидного липофусциноза нейронов.
- Диагностическая визуализация с использованием компьютерной томографии (КТ) или магнитно-резонансной томографии (МРТ). Диагностическая визуализация может помочь врачам обнаруживать изменения в головном мозге. КТ использует рентгеновские лучи и компьютер для создания детального изображения тканей и структур головного мозга и позволяет выявлять разлагающиеся или «атрофические» участки мозга у лиц с НЦЛ. МРТ использует комбинацию магнитных полей и радиоволн вместо излучения для создания изображения головного мозга.
- Измерение активности ферментов. Измерение активности тиоэстеразы пальмитоил-белка, участвующей в CLN1, кислотной протеазе, участвующей в CLN2, и, хотя более редкой, активности катепсина D, участвующей в CLN10, в лейкоцитах или культивируемых фибробластах кожи (клетки, которые укрепляют кожу и придают ей эластичность) могут быть использованы для подтверждения или исключения данных диагнозов.
- Анализ ДНК. В семьях, где подтверждена мутация в гене CLN3, анализ ДНК может быть использован для подтверждения диагноза или для пренатальной диагностики этой формы болезни Баттена. Когда мутация обнаружена, анализ ДНК также может быть использован для обнаружения незатронутых носителей этого заболевания для генетического консультирования. Если семейная мутация ранее не была идентифицирована или если общие мутации отсутствуют, последние молекулярные продвинутые исследования позволили упорядочить все известные гены NCL, увеличив шансы найти искомую мутацию (мутации).
Лечение восковидного липофусциноза нейронов
В настоящее время в Европе и США одобрен препарат Церлипоназа альфа (Cerliponase alfa) в качестве лекарственного средства для лечения медленной потери способности ходить (амбулаторной терапии) у детей в возрасте 3 лет и старше с поздней формой восковидного липофусциноза нейронов типа 2 (CLN2)(Препарат не зарегистрирован в РФ).
Не существует какого-либо конкретного лекарственного препарата, который может контролировать симптомы болезни Баттена или других НЦЛ.
Однако приступы в некоторых случаях могут быть снижены или контролироваться с помощью противосудорожных препаратов, а другие симптомы могут подвергаться рассмотрению соответствующим образом по мере их возникновения.
В то же время физическая и профессиональная терапия может помочь больным сохранить нормальное функционирование как можно дольше.
В некоторых случаях описывается замедление прогрессирования заболевания у детей с болезнью Баттена, которые лечились витаминами С и Е, а также с диетами с низким содержанием витамина А. Однако эти методы лечения не предотвращали смертельный исход заболевания.
Помощь и социальная поддержка могут позволить больным и их семьям справиться с глубокой инвалидностью и деменцией, вызванной NCL. Часто группы поддержки позволяют больным детям, взрослым и их родственникам делиться общими проблемами и опытом.
В настоящее время, учеными проводятся медицинские исследования, которые когда-нибудь могут создать эффективное лечение от этой болезни.
ЛАБОРАТОРНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ДЕТЕЙ ПЕРВЫХ 3 ЛЕТ ЖИЗНИ В НЕВРОЛОГИЧЕСКОЙ КЛИНИКЕНА ГЛАВНУЮ СТРАНИЦУ
Болезнь Янского-Бильшовского (НЛЦ 2 тип)
Причины НЦЛ 2
Заболевание обусловлено мутацией гена трипептидил пептидазы 1 (TPP1 MIM *607998). Ген картирован на коротком плече 11 хромосомы (локус 11p15). Тип наследования аутосомно-рецессивный.
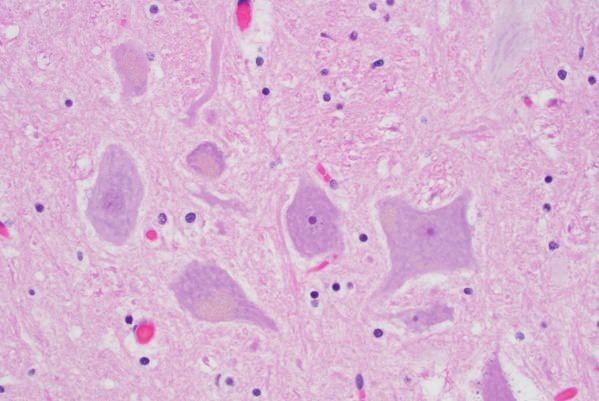
При всех формах НЦЛ происходит накопление в лизосомах клеток аутофлюоресцентного липопигмента, который состоит из белков сапозинов А и D и/или субединицы с митохондриальной АТФ синтазы. Накапливаемый материал характеризуется аутофлюоресценцией в зелено-желтом спектре при возбуждении светом длиной от 340 до 360 нм.
При гистохимическом окрашивании материал дает положительную реакцию на кислую фосфатазу и окрашивается суданом черным В, что свидетельствует о присутствии фосфолипидов. Он также является ШИК- положительным, что может указывать на высокое содержание углеводов.
Большинство из этих свойств идентично свойствам так называемых пигментов “изнашивания”, или “старения”, обычно называемых липофусцинами или цероидами.
При электронной микроскопии нейрональных и экстранейрональных тканей обнаруживают цитосомы, заполненные агрегатами характерной морфологии, описываемые как “криволинейные” профили, по типу “отпечатков пальцев”, “прямолинейные” профили, гранулярные осмиофильные отложения.
Симптомы НЦЛ 2 или болезни Янского-Бильшовского
В зависимости от возраста появления симптомов НЦЛ 2 типа подразделяют на три формы: врожденную, позднюю младенческую, юношескую формы.
Классическая форма поздней младенческой формы НЦЛ широко распространена во всем мире, но с наибольшей частотой в западной Финляндии. Обычно первые симптомы появляются в возрасте от 2х до 4х лет.
Первыми, манифестными симптомами являются генерализованные тонико-клонические приступы, задержка речевого развития, атаксия.
По мере развития заболевания присоединяются другие типы эпилептических приступов (миоклонические, парциальные, диалептические). Вслед за развитием эпилептических приступов развиваются интеллектуальные нарушения, и происходит утрата ранее приобретенных двигательных навыков.
Часто у пациентов наблюдаются атаксия, мышечная гипотония, дизартрия. Зрительные нарушения, как правило, присоединяются позднее и прогрессируют очень медленно. Мышечная гипотония сменяется спастичностью, формируются сгибательные контрактуры конечностей, формируется псевдобульбарный синдром.
В терминальную стадию болезни те же симптомы, что и при младенческой.
Диагностика НЦЛ 2
Диагностика проводится в лаборатории наследственных болезней обмена веществ МГНЦ РАМН. Основными методами подтверждения диагноза НЦЛ 2 типа являются определение активности ферментов в лейкоцитах крови или культуре клеток кожных фибробластов, молекулярно-генетические наследования, исследования биоптатов при электронной микроскопии
Лечение
В настоящее время не разработано эффективного лечения НЦЛ. Применяется симптоматическая терапия.
Подбор антиэпилептических препаратов начинают с препаратов вальпроевой кислоты. Показана высокая эффективность Ламотриджина. Препаратом выбора также являются бензодиазепины, которые не только снижают частоту эпилептических приступов, но и улучшают мышечный тонус.
- Препаратами второй очереди являются антиэпилептические препараты нового поколения: Кеппра, Трилептал, Топамакс Препараты из группы карбамазепинов и фенитоин, в некоторых случаях, способствуют учащению приступов и общему клиническому ухудшению состояния.
- Препарат триксефенидил (Тrihexyphenydil) назначают для коррекции мышечного тонуса и снижения гиперсаливации.
- Также проводится нейрометаболическая, нейротрофическая терапия (витамины группы В, Е).
Прогноз
Прогноз неблагоприятный.
Нейрональный цероидный липофусциноз 2 тип
- Нейрональный цероидный липофусциноз 2 тип.
- (НЦЛ 2, НЕДОСТАТОЧНОСТЬ ТРИПЕПТИДИЛ ПЕПТИДАЗЫ 1, КЛАССИЧЕСКИЙ ВАРИАНТ ПОЗДНЕЙ МЛАДЕНЧЕСКОЙ ФОРМЫ НЦЛ 2 ТИПА, БОЛЕЗНЬ ЯНСКОГО-БИЛЬШОВСКОГО, сLINCL).
- CLN 2; CEROID LIPOFUSCINOSIS, NEURONAL, 2; CLN2; CEROID LIPOFUSCINOSIS, NEURONAL, 2, VARIABLE AGE AT ONSETJAN SKY-BIELSCHOWSKY DISEASE; NEURONAL CEROID LIPOFUSCINOSIS, LATE INFANTILE, INCLUDED; LINCL, INCLUDED
- MIM#204500
Генетика: мутации гена трипептидил пептидазы 1 (TPP1 MIM *607998). Ген картирован на коротком плече 11 хромосомы (локус 11p15).
Эпидемиология:суммарная частота встречаемости всех форм НЦЛ в мире составляет 1: 25000.
Тип наследования:аутосомно-рецессивный
Патогенез:при всех формах НЦЛ происходит накопление в лизосомах клеток аутофлюоресцентного липопигмента, который состоит из белков сапозинов А и D и/или субединицы с митохондриальной АТФ синтазы.
Накапливаемый материал характеризуется аутофлюоресценцией в зелено-желтом спектре при возбуждении светом длиной от 340 до 360 нм. При гистохимическом окрашивании материал дает положительную реакцию на кислую фосфатазу и окрашивается суданом черным В, что свидетельствует о присутствии фосфолипидов.
Он также является ШИК- положительным, что может указывать на высокое содержание углеводов. Большинство из этих свойств идентично свойствам так называемых пигментов “изнашивания”, или “старения”, обычно называемых липофусцинами или цероидами.
При электронной микроскопии нейрональных и экстранейрональных тканей обнаруживают цитосомы, заполненные агрегатами характерной морфологии, описываемые как “криволинейные” профили, по типу “отпечатков пальцев”, “прямолинейные” профили, гранулярные осмиофильные отложения.
Клинические проявления:в зависимости от возраста манифестации НЦЛ 2 типа подразделяют на три формы: врожденную, позднюю младенческую, юношескую формы. Классическая форма поздней младенческой формы НЦЛ широко распространена во всем мире, но с наибольшей частотой в западной Финляндии. Обычно первые симптомы появляются в возрасте от двух до четырех лет.
Манифестными симптомами являются генерализованные тонико-клонические приступы, задержка речевого развития, атаксия. По мере развития заболевания присоединяются другие типы эпилептических приступов (миоклонические, парциальные, диалептические).
Вслед за развитием эпилептических приступов развиваются интеллектуальные нарушения, и происходит утрата ранее приобретенных двигательных навыков. Часто у пациентов наблюдаются атаксия, мышечная гипотония, дизартрия. Зрительные нарушения, как правило, присоединяются позднее и прогрессируют очень медленно.
Мышечная гипотония сменяется спастичностью, формируются сгибательные контрактуры конечностей, формируется псевдобульбарный синдром. В терминальную стадию болезни те же симптомы, что и при младенческой
Диагностика:основными методами подтверждения диагноза НЦЛ 2 типа являются определение активности ферментов в лейкоцитах крови или культуре клеток кожных фибробластов, молекулярно-генетические наследования, исследования биоптатов при электронной микроскопии
Лечение: в настоящее время не разработано эффективного лечения НЦЛ. Применяется симптоматическая терапия. Подбор антиэпилептических препаратов начинают с препаратов вальпроевой кислоты. Показана высокая эффективность Ламотриджина.
Препаратом выбора также являются бензодиазепины, которые не только снижают частоту эпилептических приступов, но и улучшают мышечный тонус.
Препаратами второй очереди являются антиэпилептические препараты нового поколения: Кеппра, Трилептал, Топамакс Препараты из группы карбамазепинов и фенитоин, в некоторых случаях, способствуют учащению приступов и общему клиническому ухудшению состояния.
Препарат триксефенидил (Тrihexyphenydil) назначают для коррекции мышечного тонуса и снижения гиперсаливации. Также проводится нейрометаболическая, нейротрофическая терапия (витамины группы В, Е). Диагностика проводится в лаборатории наследственных болезней обмена веществ МГНЦ РАМН (http://www.labnbo.narod.ru).
Прогноз
Прогноз неблагоприятный.
Болезнь Баттена
Болезнь Баттена (НЦЛ) – это редкое нейродегенеративное рецессивно наследуемое заболевание, которое входит в группу восковидных липофусцинозов нейронов, проявляется в детстве и приводит к смерти.
- Симптомы
- Диагностика
- Лечение
- Прогноз
При данном заболевании жировые вещества скапливаются в клетках нервной системы. Минимум восемь генов были обнаружены в связи с этой болезнью, однако наиболее распространенная форма развивается по причине мутаций в гене CLN3.
Болезнь Баттена названа в честь Фредерика Баттена, педиатра, который впервые описал данный недуг в 1903 году.
Симптомы заболевания практически всегда возникают в детстве или подростковом возрасте (5−15 лет). Болезнь Баттена имеет следующие признаки:
- глаза окутаны пеленой (возникает ощущение, что наступает слепота);
- эпилептические приступы;
- болезненные ощущения в голове;
- припадки бешенства;
- дегенеративные изменения;
- изменения в поведении.
Иногда симптомы очень слабые, поэтому не ощутимы для ребенка и не видны для окружающих. В данном случае необходим еженедельный осмотр при помощи специальных устройств и учет у врача. Время проявления определенных симптомов, тяжесть и скорость их прогрессирования находятся в зависимости от того, к какому типу относится болезнь Баттена.
При определении заболевания большую роль играет обнаружение вакуолизированных лимфоцитов в периферической крови, а также дегенерация сетчатки.
В разных тканях пациентов обнаруживается пигмент, которых схож с цероидом и липофусцином. При проведении электронной микроскопии можно заметить, что пигмент по внешнему виду напоминает отпечатки пальцев и криволинейных контуров.
На сегодняшний день ферментный дефект, который лежит в основе болезни, неизвестен.
В настоящее время лечение от данного заболевания не найдено. Некоторые медикаментозные средства позволяют только временно снять симптомы.
В ноябре 2006 года, когда было получено одобрение из Управления по контролю качества лекарств и продуктов, Орегонский университет здоровья и науки начал клиническое исследование.
При этом очищенные нервные стволовые клетки ввели в мозг ребенка шести лет, который страдал НЦЛ и разучился говорить. Это было первое введение фетальных стволовых клеток в мозг пациента.
Уже в начале декабря ребенок, у которого был установлен диагноз «болезнь Баттена», почувствовал заметное облегчение. Он смог вернуться домой, появилась речь.
Главная цель этапа состояла в изучении безопасной трансплантации. И она была установлена, так как шесть больных, которые в рамках исследования получили лечение, а также иммуносупрессию, перенесли терапию хорошо. В результате их нейропсихологическое, медицинское и неврологическое состояние находилось в соответствии с нормальным течением заболевания.
Болезнь Баттена имеет неблагоприятный прогноз. Заболевание приводит к смерти.
nclfamilies.ru: о болезни
Нейрональный цероидный липофусциноз (НЦЛ) относится к обширному классу наследственных заболеваний обмена веществ, под названием лизосомные болезни накопления (ЛБН). Это достаточно редкое заболевание, которое только в конце прошлого века включили в класс ЛБН.
На основании возраста начала болезни, скорости прогрессирования, нейрофизиологических и морфологических данных, выделяют инфантильную (НЦЛ1), позднюю инфантильную (НЦЛ2), ювенильную (НЦЛ3) и взрослую формы заболевания, а так же довольно большое количество атипичных клинических форм.
Суммарная частота встречаемости всех форм НЦЛ в мире составляет 1:25000.
В США и некоторых странах Европы все типы нейронального цероидного липофусциноза объединили под общим названием – болезнь Баттена.
Это заболевание в основном затрагивает детей и подростков, однако есть редкие формы НЦЛ, проявляющиеся у взрослых. В зависимости от типа или формы НЦЛ, это заболевание может проявляться в различное время развития ребенка или подростка. Так НЦЛ 1 дает о себе знать к концу первого года жизни малыша, НЦЛ 2 –примерно в 2-4 года, НЦЛ3- 8-10лет.
Как правило, такие дети рождаются здоровыми и в первые годы своей жизни, до начала заболевания, достигают многих успехов в развитии. Они хорошо развиты физически, контактны, посещают детские учреждения, психологически развиты по возрасту.
Однако может наблюдаться небольшое отставание в развитии речи или некоторых навыков. НЦЛ может проявляться постепенно, начало заболевания определяется одним из трех признаков: судороги, атаксия (неуклюжесть походки) и снижение зрения.
Эти симптомы могут быть при очень многих других заболеваниях, а болезнь Баттена настолько редко встречается, что почти всегда её диагностируют не сразу. То есть сначала ребенку ставят неправильный основной диагноз, как правило, эпилепсию или какое-либо заболевание глаз.
Тем не менее, по мере прогрессирования заболевания вскоре становится очевидно, что первоначальный диагноз неверен и начинается более серьезное обследование больного ребенка.
Прогрессирование заболевания заключается в ухудшении зрения, потери навыков, речи, потери способности стоять и ходить самостоятельно.
Как правило, сначала ребенку делают МРТ головного мозга и по данным такого исследования опытный врач-невролог может предположить наличие заболевания.
Если данные МРТ (атрофия больших полушарий и мозжечка) позволяют сделать выводы о наличии заболевания, ребенка и его родителей отправляют на генетическую экспертизу.
НЦЛ является генетическим заболеванием. Различные формы его вызваны дефектами (мутациями) в различных генах NCL. НЦЛ-генетическое прогрессирующее заболевание головного мозга детей и подростков. NCL заболевания, которые встречаются в зрелом возрасте, очень редки и четко не определены.
В случае НЦЛ 2 типа из-за наличия мутантного гена NCL2 снижено содержание фермента TPP1 (трипептидил пептидазы 1) или же фермент отсутствует вообще (TPP1-лизосомный фермент).
Как следствие, не разрушается липофусцин и образуется цероид в мозге. Следует отметить, что эти включения могут накапливаться не только в нейронах, но и в лимфоцитах, биоптатах кожи, конъюктивы, слизистой прямой кишки и клетках сетчатки, сердечной мышце, печени.
Однако, при НЦЛ это происходит только в нейронах и почему- до сих пор неизвестно… Под действием липофусцина погибают нейроны, что приводит к атрофии больших полушарий мозга и мозжечка. Это хорошо видно на снимках МРТ.
И именно данные МРТ наряду с клиническими проявлениями позволяют врачам предположить наличие болезни.
Подтверждается заболевание данными генетического анализа крови ребенка и его родителей. Тип наследования этого заболевания аутосомно-рецессивный, это означает, что здоровые родители являются носителями нормального и дефектного гена NCL и вероятность наследования дефектных генов в будущем ребенке составляет 25%.
В организме больного ребенка нет нормального гена NCL, отвечающего за продуцирование фермента ТРР1, который в свою очередь трудится над очищением нейронов от токсического пигмента липофусцина (или родственного ему). Таким образом, мозг засоряется этим мусором и гибнут нейроны. Обычно первые симптомы НЦЛ2 проявляются в возрасте 2-4 лет (чаще в 3 года).
До начала заболевания ребенок ,как правило, ничем не отличается от своих сверстников. Манифестными симптомами являются эпилептические приступы, задержка речевого развития, атаксия.
По мере развития заболевания наблюдается прогрессирующая атрофия головного мозга, с чем связаны интеллектуальные нарушения, утрата ранее приобретенных навыков, постепенное снижение зрения, слепота, полная обездвиженность, нарушение приема пищи, кома. В конце концов, дети с болезнью Баттена становятся слепыми, прикованными к постели и не могут общаться.
Это заболевание всегда заканчивается смертельным исходом. Болезнь не проходит у двух детей одинаково. В общем, можно сказать, что все больные дети будут иметь судороги, все будут терять зрение и т.д. Однако нельзя предсказать, когда именно это будет происходить с каждым отдельным ребенком с НЦЛ.
Можно сказать, что каждый заболевший ребенок имеет своё индивидуальное поведение в болезни. Заболевание является нейродегенеративным, страдают нейроны головного мозга. И поскольку, мозг отвечает за работу всего организма, то со временем органы и системы организма перестают работать, что приводит к летальному исходу. На сегодняшний день эта болезнь неизлечима.
Ведется симптоматическое лечение. Посредством корректирующих препаратов удается облегчить состояние больного. К этим препаратам относятся противоэпилептические, миорелаксанты, обезболивающие, седативные, антипсихотические, витамины гр. В, вит. Е, L-карнитин и др. Какие-либо методы физиотерапии приносят временное улучшение, а иногда даже и вредят.
В некоторых странах идут активные поиски терапии НЦЛ. Тем, что принесет излечение или замедлит развитие болезни, может быть лекарственный препарат, метод замены больного гена на здоровый или метод повышение продуцирования фермента. Рассматриваются методы терапии стволовыми клетками. В клинических лабораториях идут исследования на животных. В одном из университетов США в исследованиях принимают участие добровольцы, больные НЦЛ.
- Результаты, в некотором смысле, обнадеживающие и остается ждать и надеется, что лечение от этого разрушающего заболевания будет найдено в скором времени.
- АУТОСОМНО-РЕЦЕССИВНЫЙ ТИП НАСЛЕДОВАНИЯ
Наследственные заболевания- это болезни, обусловленные нарушениями в процессах хранения, передачи и реализации генетической информации.
С развитием генетики человека, в том числе и генетики медицинской, выяснилась наследственная природа многих заболеваний и синдромов, считавшихся ранее болезнями с неустановленной этиологией.
Роль наследственных факторов подтверждается более высокой частотой ряда заболеваний в некоторых семьях по сравнению с населением в целом.
В основе наследственных заболеваний лежат мутации — преимущественно хромосомные и генные, соответственно чему условно говорят о хромосомных болезнях и собственно наследственных (генных) болезнях.
Мутация ведёт к нарушению синтеза определенного полипептида (структурного белка или фермента).
В зависимости от того, какова роль этого полипептида в жизнедеятельности организма, у больного возникают нарушения (изменения фенотипа) локального или системного порядка.
Наследственно дегенеративные болезни отличаются деструктивными и дистрофическими изменениями в тканях (нервной, мышечной и др.), избирательностью поражения нервной системы, мышц, внутренних органов и кожи, прогрессирующим течением. Одни из них возникают с первых лет жизни, другие — много лет спустя после рождения.
Типы наследования различны: аутосомно-доминантный, аутосомно-рецессивный и рецессивный, сцепленный с полом.
В зависимости от того, где локализован патологический (мутантный) ген — в аутосоме или в половой хромосоме — и каковы его взаимоотношения с нормальным аллелем, т. е.
является ли мутация доминантной (нормальный ген подавляется патологическим) или рецессивной (патологический ген подавляется нормальным) и различают типы наследования.
При заболеваниях, наследуемых по аутосомно-рецессивному типу, мутантный ген проявляется лишь в гомозиготном состоянии; больные мальчики и девочки рождаются с одинаковой частотой; родители больных фенотипически здоровы, но являются гетерозиготными носителями мутантного гена; патологическая наследственность прослеживается в родословной семьи “по горизонтали”; вероятность рождения больных детей возрастает в случае кровного родства родителей. Если один из родителей гомозиготен по патологическому рецессивному гену, а другой является его гетерозиготным носителем, то в половине случаев дети могут оказаться больными, и создаётся впечатление наследования заболевания по доминантному типу. Такое явление носит название псевдодоминирования. От истинного доминирования оно отличается тем, что больные с рецессивной мутацией в браке со здоровыми людьми всегда будут давать здоровое потомство, а здоровые в браке с гетерозиготными носителями с определенной частотой (25%) будут иметь больных детей.
Что такое болезнь Баттена. Болезнь Баттена (Вогта-Шпильмейера-Шагрена или NCL 3)
Причина возникновения недуга – наследственное нарушение строения ДНК, которое передаётся по мужской и женской линиям. При этом у больного ребёнка нет гена NCL, отвечающего за выработку вещества, которое необходимо для переработки токсического липофусцина, который является жёлтым пигментом и вырабатывается при разложении отработавших митохондрий.
Липофусцин также называют пигментом старения или изнашивания. Повышенное содержание данного вещества наблюдается в старости или при заболеваниях с деградацией тканей.
В идеале, оно должно быть переработано митохондриями при помощи специального фермента TPP1, но так как гена, отвечающего за его продуцирование нет, то липофусцин накапливается как токсинный мусор в различных клетках и разрушает их (при болезни Баттена процесс накопления происходит в нейронах).
Из-за массовой гибели нейронов происходит атрофия мозжечка, а также больших полушарий, что хорошо различимо во время проведения магнитно-резонансной томографии.
Так как заболевание наследственное, то практически всегда проявляется в детском возрасте, при этом конкретный возраст появления первых симптомов зависит от скорости выработки липофусцина, поэтому, чем медленнее она происходит, тем позже проявляется заболевание и тем дольше жизнь больного с момента диагностики недуга (не считая атипичных случаев).
Симптомы
До накопления липофусцина в нейронах головного мозга больные дети не отличаются от своих сверстников.
По достижению количества этого вещества определённого порога, когда начинается гибель нейронов головного мозга начинают наблюдаться первые симптомы в виде отставания в развитии, эпилептических приступов, а также атаксии.
Атаксия – нарушение координаций движений с проявлениями мышечной слабости, вызванное поражением мозжечка, коры мозга или вестибулярного аппарата.
С прогрессией атрофии мозга начинаются умственные отклонения, потеря навыков, потеря зрения.
В конечном итоге заболевание приводит к полной потере мозговых функций сначала в виде обездвиженности, затем в отсутствии элементарных рефлексов и функций внутренних органов. Конечной стадией заболевания является кома, после которой наступает смерть из-за гибели головного мозга.
Окулогирный криз от нейролептика: лечение, симптомы
Так как гибель нейронов происходит неравномерно и у каждого ребёнка индивидуально, то качество проявлений симптомов, их последовательность и промежутки между стадиями могут быть у каждого разными.
Диагностика заболевания
Болезнь Баттена диагностируется прежде всего при помощи анализа ДНК, а также некоторых методов обследования:
- Общие анализы (вакуолизированные лимфоциты в крови).
- Осмотр у окулиста (дегенерация сетчатки).
- ЭЭГ (нарушение его нормальной электрической активности).
- МРТ мозга (его дегенеративные изменения).
- Биопсия тканей кожи и других (наличие в клетках сходного вещества с липофусином).
Лечение
На сегодняшний день это неизлечимое заболевание, так как пока не найдено абсолютно каких методов, способных хоть как-то остановить или замедлить процесс деградации нейронов. Единственное, что можно сделать максимально облегчить и продлить последние дни больного при помощи симптоматической и интенсивной терапии.
Прогноз болезни
Люди, страдающие от болезни Баттена, могут стать полностью слепыми, прикованными к постели и неспособными передвигаться. В основном патология приводит к летальному исходу, и сильно сокращает продолжительность жизни. Однако при проведении правильно подобранной терапии некоторые пациенты живут до 30 лет.
Болезнь Баттена. Симптомы и проявления
Симптомы и проявления варьируются у каждого ребенка. Ранние симптомы болезни Баттена сбивают с толку и их не легко распознать, даже опытному медицинского персоналу. Ниже приведено краткое описание наиболее типичной симптоматики:
- Нарушения зрения часто прогрессируют до полной слепоты
- Приступы, они могут быть частыми и их трудно контролировать
- Снижение когнитивных функций
- Личностные и поведенческие изменения
- Потеря навыков общения
- Потеря мелкой и крупной моторики
- Аномальные телодвижения
- Общее прогрессирующее ухудшение
Другие симптомы, которые могут развиться, включают в себя: замедление роста головы с возрастом в инфантильной форме, плохое кровообращение в нижних конечностях, ноги и ступни будут холодными, ожирение, искривление позвоночника, гипервентиляция и / или задержки дыхания, затрудненное глотание и проблемы с кормлением, скрежетание зубами и запоры.
Классификация
Современной медицине известно четыре формы заболевания. Они имеют схожую симптоматику, но отличаются скоростью прогрессирования и возрастом пациентов.
Детская форма болезни Баттена проявляется до исполнения ребенку 2 лет и быстро развивается. Заболевание тормозит общий рост, а в качестве осложнений появляются миоклонические судороги и микроцефалия. В 95% случаев ребенок умирает в возрасте до 5 лет, в остальных 10% смерть наступает в возрасте 6–8 лет.
Поздняя форма проявляется у детей 2–4 лет. Заболевание сопровождается частыми судорогами и мышечной слабостью. Прием противосудорожных препаратов не приносит облегчения. В результате быстрого прогрессирования заболевания летальный исход наступает в возрасте 6–12 лет.
Ювенальная форма болезни Баттена диагностируется в возрастном промежутке с 5 до 12 лет. Характерные симптомы патологии: судороги, потеря зрения и задержка развития (умственного и физического). В течение длительного времени сохраняется нормальная двигательная активность пациента. Развивается болезнь медленно, и пациент, как правило, доживает до тридцатилетнего возраста.
Взрослая форма встречается крайне редко и диагностируется у людей старше 40 лет. Прогрессирует заболевание медленно, а симптомы малозаметны. Летальный исход наступает через 10–15 лет после диагностирования болезни Баттена.
Болезнь Баттена. Эпидемиология
Болезнь Баттена и другие ее формы встречаются относительно редко. По частоте развития, имеется следующая статистика:
- 2-3 из каждых 100000 родившихся в Соединенных Штатах
- 1 из каждых 12500 родившихся в Финляндии
- 1 из каждых 20000 родившихся в Норвегии и Швеции
- 1 из каждых 30000 – 40000 родившихся в Великобритании
- Другая информация отсутствует
Симптомы болезни Баттена
Прогрессирующая потеря зрения от 4-10 лет, к более подростковому возрасту может сформироваться полная слепота.
При поражении зрения на первый план выходят такие симптомы как: ночная слепота, туннельное зрение (отсутствует периферийное зрение) или наоборот – периферийное зрение (отсутствует центральное зрение), сеточное зрение, антипатия к яркому свету, медленная регулировка от темного к светлому, и наоборот, размытые очертания видимых объеков – т.е нечёткость зрения, плохое различение цветов.
Черепно-мозговые нервы, 12 пар черепных нервов в таблице
Вариабельность клинических проявлений зависит от степени поражения нервной ткани.
Что касаемо психоневрологических проявлений, то тут нужно говорить о постепенной утрате социальной адаптации: психомоторная дегенерация (спокойствие, вялость могут быстро сменяться судорожными припадками, отмечается частая и резкая смена настроения), деменция – стойкое снижение познавательной деятельности с утратой приобретённых навыков (в том числе ходьбы, сидения, простых и необходимых элементов самообслуживания), высокая судорожная готовность, утрата речевых навыков в следствии поражения центрального речедвигательного аппарата (понимание услышанной речи остаётся).
Основные причины
Болезнь Баттена провоцируется нарушениями в генах, связанных с выработкой некоторых белков организма. Заболевание приводит к постепенному накоплению жиров и белков в клетках глаз, головного мозга, кожи.
Ученым удалось определить дефектные ферменты в мутировавших генах, что позволяет выявить протекание патологии на первоначальном этапе и провести лечение. Кроме того, это имеет важное значение в профилактике.
Существуют определенные факторы риска, которые значительно повышают вероятность развития болезни. К ним нужно отнести генетическую предрасположенность. Кроме того, от болезни страдают дети тех родителей, которые сами не болеют, но при этом являются носителями дефектного гена.
Проведение диагностики
Диагностика заболеваний нервной системы вызывает определенные сложности, так как их симптомы часто схожи с проявлениями других патологических процессов в организме. Первоначальный диагноз устанавливается при осмотре глазного дна. Для подтверждения поставленного диагноза назначаются:
- анализ крови;
- анализ мочи;
- биопсия тканей.
Чтобы определить наличие аномалий головного мозга, требуются и определенные виды исследований:
- сканирование МРТ;
- компьютерная томография;
- электроэнцефалография.
Для подтверждения поставленного диагноза назначают электрофизиологические методы исследования, чтобы можно было обнаружить имеющиеся проблемы со зрением, связанные с протеканием болезни Баттена. Чтобы выявить отклонения, которые провоцируют заболевание, назначается анализ ДНК.
- http://NashiNervy.ru/o-nervnoj-sisteme/chto-takoe-bolezn-battena.html
- http://fb.ru/article/421070/bolezn-battena-vozmojnyie-prichinyi-simptomyi-variantyi-lecheniya
- http://redkie-bolezni.com/bolezn-battena/
- https://dolgojit.net/bolezn-battena.php
- https://medicalj.ru/diseases/congenital-anomaly/1401-bolezn-battena
Comments
(0 Comments)