Болезнь пика: причины, симптомы и диагностика, лечение у детей
Болезнь Пика (Ниманна-Пика) является семейным заболеванием, передающимся по наследству. Патология проявляется избыточным накоплением жировой ткани во внутренних органах, включая и головной мозг. Болезнь Пика различается несколькими клиническими формами.
Причины и особенности заболевания
Болезнь Пика развивается в результате того, что определенные ферменты недостаточно активны, и это вызывает накопление продуктов обмена веществ, в данном случае жиров. В норме избыток жиров расщепляется и выводится из организма, чего не происходит у больных этим заболеванием.
Различают четыре формы этого заболевания – тип А, В, С, D. Каждая форма болезни Ниманна Пика провоцируется мутацией в определенном наборе хромосом.
Дефект может наблюдаться в 11-ой, 14-ой или 18-ой хромосоме.
Результатом такого отклонения от нормы становится нарушение расщепления сфингомиелина, в результате чего происходит нарушение обмена веществ и накопление жиров в тканях внутренних органов.
Способ наследования
Патология наследуется по аутосомно-рецессивному типу. Таким образом, если один из родителей болен, ребенок унаследует патологию, независимо от пола.
Наиболее неблагоприятным считается случай, если оба родителя являются носителями патологического гена. В этом случае болезнь Пика у ребенка будет протекать тяжелее. Как правило, прогноз при этом неблагоприятный.
Типы заболевания
Симптоматика, характер развития и прогноз во многом зависят от типа заболевания. Различают четыре типа болезни Ниманна Пика:
- классическая форма (тип А);
- висцеральный тип (тип В);
- ювенильная форма (тип С);
- особая форма Новой Скотии (тип D).

Классическая форма характеризуется острым нейропатическим развитием. При болезни Ниманна Пика висцерального типа наблюдается хроническое течение, однако нервная система не вовлечена в патологический процесс.
Ювенильная форма Болезни Пика тип С отличается подострым характером развития с вовлечением нервной системы.
Тип D встречается только среди жителей Новой Скотии (Канада), поэтому во многих источниках его объединяют с ювенильной формой из-за схожести симптомов.
Симптомы патологии
Для каждой формы болезни Ниманна Пика характерны свои симптомы и особенности развития.
Классическая форма патологии считается наиболее неблагоприятной. Дети рождаются абсолютно здоровыми, однако патология быстро прогрессирует в течении первых недель жизни. Затем появляются следующие симптомы:
- отсутствие аппетита;
- постоянная тошнота до рвоты;
- отставание в росте, стремительное снижение веса;
- увеличение размеров печени;
- увеличение объема селезенки;
- пигментация кожи;
- увеличение лимфоузлов;
- помутнение роговицы глаза.
Живот пациента выглядит очень большим из-за увеличения размеров внутренних органов.
Следующим этапом развития болезни становится поражение центральной нервной системы, что проявляется нарушением развития, отсутствием речи, нарушениями слуха и зрения. Рефлекторная активность пациентов сильно повышена. Возможно развитие судорожных припадков, как при эпилепсии.
Эта форма патологии является наиболее тяжелой, летальный исход наступает в возрасте от одного года до пяти лет. Причиной смерти, как правило, становится истощение.
Второй тип заболевания (тип В) отличается благоприятным течением. При такой форме болезни Пика симптомы следующие:
- увеличение размеров селезенки;
- увеличение размеров печени;
- нарушение свертываемости крови;
- анемия;
- нарушение пищеварения.
Увеличение размеров внутренних органов диагностируется на шестом году жизни ребенка. Увеличение печени приводит к нарушению кровообразования, результатом чего часто становится анемия.
Внешние проявления выражаются в увеличении размеров живота, однако патология не так заметна, как при первом типе заболевания. Пациентов часто беспокоит тошнота и рвота, боли в желудке и запоры.
Больные этой формой патологии часто простужаются.
- В этом случае не наблюдается поражения нервной системы, что обуславливает долгий срок жизни пациентов.
-

- Третий тип патологии (тип С) нередко проявляется в подростковом возрасте до 20 лет. Для этой формы характерны следующие признаки:
- незначительно увеличение внутренних органов;
- нарушение зрения;
- пигментация кожи;
- снижение мышечного тонуса;
- спастические парезы;
- нарушение вращения глазного яблока;
- изменение походки;
- тремор пальцев;
- прогрессирование умственной отсталости;
- нарушение функции глотания.
Со временем к симптомам присоединяется слабость мышц ног, из-за чего пациент не может ходить. Прогноз при болезни Пика тип С во многом зависит от симптомов, наблюдающихся у пациента. Летальный исход быстро наступает при наличии множества симптомов. Болезнь Пика типа Д имеет такие же симптомы, однако отличается процессом развития.
Лечение патологии

Вылечить патологию невозможно. Медикаментозное лечение направлено на ослабление симптомов. С этой целью применяют:
- препараты против судорожных припадков;
- антидепрессанты;
- средства против диареи и для улучшения функции ЖКТ;
- препараты против мышечных спазмов и дрожания мышц;
- антибиотики и противопростудные лекарства.
Для замедления прогрессирования симптомов поражения нервной системы больным показан препарат Миглустат. Терапия этим лекарственным средством позволяет остановить прогрессирование болезни и продлить жизнь пациенту на долгие годы. Положительный эффект наблюдается только при длительном приеме лекарства, поэтому терапевтический курс длится не менее полугода.
Для поддержания функции пораженных органов назначается переливание крови. Для поддержки жизнедеятельности организма рекомендованы инъекции витаминных препаратов.
Прогноз
Синдром Пика-Ниманна является неизлечимым заболеванием, которое приводит к летальному исходу. Наиболее тяжелой формой патологии является первый тип, в этом случае летальный исход наступает в течении первых лет жизни.

В остальных случаях пациенты могут дожить до 30 лет. Прогноз при этом зависит от лечения и применяемых терапевтических методов. Развитие заболевания происходит стремительно. Пик развития патологии приходится на период поражения нервной системы. В это время наблюдается стремительный регресс психического развития пациента, потеря слуха, ухудшение зрения.
Профилактических мер при болезни Ниманна Пика не существует. Семьям, родственники которых больны этим синдромом, необходимо пройти обследование у генетика, перед планированием беременности.
Болезнь Нимана-Пика: что это такое, симптомы и лечение
Болезнь Нимана-Пика (сфингомиелиноз) – это наследственное заболевание, связанное с избыточным накоплением жиров в различных органах и тканях, в первую очередь в головном мозге, печени, лимфатических узлах, селезенке, костном мозге. Имеет несколько клинических вариантов, у каждого из которых различный прогноз. Специфического лечения в настоящее время нет. Из этой статьи Вы сможете узнать о причине, симптомах и возможностях лечения болезни Нимана-Пика.
Болезнь Нимана-Пика относится к лизосомальным болезням накопления. Это когда в результате недостаточной активности какого-либо фермента в клетках организма накапливаются промежуточные продукты обмена веществ, которые в норме подвергаются дальнейшему расщеплению.
Причины болезни Нимана-Пика
В основе заболевания лежит генетический дефект 11-й хромосомы (типы А и В), 14-й и 18-й хромосомы (тип С).
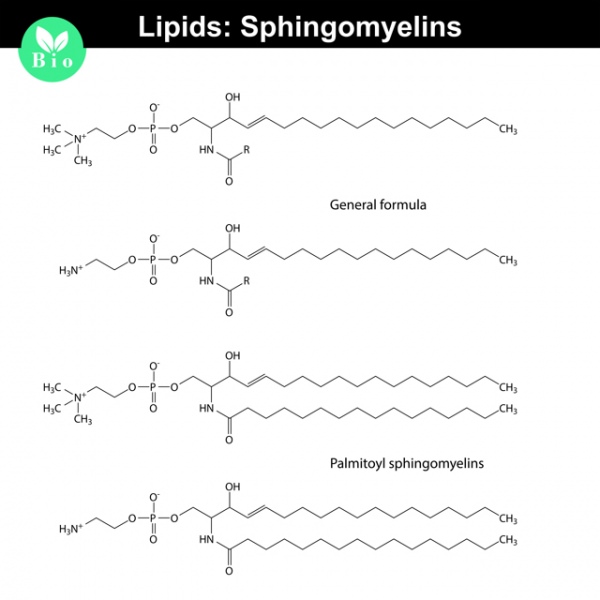
В результате наличия нарушения в структуре гена у человека наблюдается снижение активности фермента сфингомиелиназы, которая расщепляет сфингомиелин. Сфингомиелин – это разновидность жира.
Такое биохимическое нарушение приводит к избыточному накоплению сфингомиелина и холестерина в клетках ретикуло-эндотелиальной системы: тканевых макрофагах. В результате нарушается обмен веществ.
Тканевые макрофаги разбросаны по всему организму, но больше всего их в селезенке, печени, костном мозге, лимфатических узлах, центральной нервной системе.
Заболевание носит аутосомно-рецессивный характер, то есть оно не связано с полом, могут болеть как мужчины, так и женщины. При совпадении двух патологических генов (от отца и от матери) недуг протекает наиболее тяжело.
Симптомы
Выделяют несколько клинических вариантов болезни Нимана-Пика. Разделение на варианты обусловлено особенностями течения и биохимическими изменениями.
Всего изучено 4 типа болезни:
- тип А – классическая форма болезни (инфантильная, острая нейропатическая);
- тип В – висцеральная форма (хроническая, без вовлечения нервной системы);
- тип С – ювенильная форма (подострая, хроническая нейропатическая);
- тип D – форма из Новой Скотии (по названию провинции в Канаде, у жителей которой встречается эта форма). В последнее время этот тип объединили с типом С.
Тип А
Это наиболее неблагоприятная в отношении прогноза для жизни форма. Проявляет себя через несколько недель после рождения (при рождении дети выглядят здоровыми).
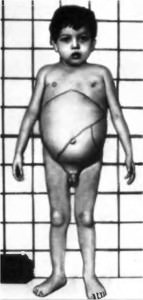
У ребенка ухудшается аппетит, он начинает терять в весе и отставать в росте. Возможны периодические рвоты и поносы. Постепенно увеличивается живот из-за печени и селезенки (печень увеличивается раньше, чем селезенка), развивается асцит.
Конечности выглядят тонкими и очень худыми по сравнению с увеличенным животом.
Кожа ребенка становится сухой, теряет свою эластичность, приобретает желтоватый цвет, местами определяются желтовато-серые или желто-коричневые пятна. Увеличиваются все группы лимфатических узлов, что можно определить при прощупывании (пальпации).
При осмотре глазного дна определяется специфический симптом «вишневой косточки» — темно-красного цвета пятнышко на сетчатке. Возможно помутнение роговицы и появление коричневой окраски хрусталика.
Поражение нервной системы заключается вначале в отставании в нервно-психическом развитии от сверстников: детки не держат голову, не переворачиваются с живота на спину, не следят за игрушкой. Повышается мышечный тонус в руках и ногах, развивается мышечная слабость.
Сухожильные рефлексы также повышаются. Постепенно утрачивается слух, снижается зрение, могут быть эпилептические припадки.
В разгаре заболевания ребенок вял и апатичен, слабо реагирует на происходящие вокруг него события, практически постоянно пребывает с открытым ртом, из-за чего развивается слюнотечение.
Возникают периоды внезапного повышения температуры: гипертермические кризы.
Постепенно развивается истощение, и больные этой формой заболевания погибают в возрасте 2-4 лет.
Тип В
Это форма болезни имеет благоприятное течение. В этом случае нервная система не поражается, накопление сфингомиелина и холестерина происходит только во внутренних органах. Почему нервная система остается нетронутой — до сих пор остается загадкой для врачей.
Вначале увеличивается селезенка, обычно это происходит к 2-6 годам. Позже увеличивается печень. Поражение печени приводит к повышенной кровоточивости из-за нарушения свертывающей системы крови. Часто развивается анемия. Беспокоят боли в животе, периодические нарушения стула, изредка тошнота и рвота. Живот увеличивается в размерах, но не так значительно, как при типе А.
В связи с накоплением жиров в легочной ткани формируются инфильтраты. Это обусловливает частые простудные заболевания у таких детей.
Эта форма характеризуется длительным хроническим течением. Продолжительность жизни значительно дольше, чем при типе А, больные доживают до взрослого возраста.
Тип С
Биохимический дефект при этой форме точно не выяснен. Предполагается нарушение транспорта сфингомиелина. Наблюдается нерезкое накопление сфингомиелина и значительное — холестерина в головном мозге, селезенке и печени.
Заболевание впервые проявляет себя в промежутке от 2 до 20 лет. Увеличение печени и селезенки по сравнению с типами А и В незначительное. Характерен желтушный оттенок кожи. На глазном дне – симптом «вишневой косточки», пигментная дегенерация сетчатки.
Неврологические нарушения начинаются со снижения мышечного тонуса, который затем, наоборот, повышается. Постепенно формируются спастические парезы: слабость мышц с одновременным повышением мышечного тонуса. Нарушается совместная деятельность глазных яблок, становятся невозможными согласованные движения глаз, особенно при взгляде вверх (так называемый вертикальный офтальмопарез).
Развивается нарушение координации, в связи с чем меняется походка. Присоединяются дрожание и непроизвольные движения в конечностях. Характерны насильственные выкручивающие движения в голове и туловище (торсионная дистония). Появляются эпилептические припадки. Нарушается глотание и речь.
Умственные нарушения постепенно прогрессируют, дети утрачивают способность к обучению, в конце концов развивается деменция (слабоумие). Нарушается контроль над функцией тазовых органов.
Описан довольно специфичный симптом для данной формы болезни Нимана-Пика: это внезапная потеря мышечного тонуса в ногах, челюсти и шее при смехе или других сильных эмоциях. Заболевание постепенно прогрессирует.
После появления развернутой клинической картины заболевания дни таких больных сочтены.
Тип D
Описан среди жителей провинции Канады: Новой Шотландии (Скотии). Четкий биохимический дефект не выявлен, но заболевание развивается в результате небольшого накопления сфингомиелина и значительного — холестерина. По своим клиническим проявлениям практически не отличается от типа С, поэтому некоторые исследователи предпочитают не выделять ее в отдельную форму.
Диагностика
-
- Для подтверждения диагноза определяют активность сфингомиелиназы в культуре фибробластов кожи и лейкоцитах (для типа А и В), обнаруживают накопление неэтерифицированного холестерина в культуре фибробластов кожи (для типа С), проводят поиск генетических дефектов в 11, 14, 18-й хромосомах.
- Пункция костного мозга у таких больных обнаруживает специфические «пенистые» клетки Нимана-Пика (они так выглядят из-за накопления жиров).
Лечение
Заболевание неизлечимо. В основном проводится симптоматическое лечение для облегчения страданий больного.
Среди симптоматических средств применяют:
- противосудорожные (Депакин и другие Вальпроаты);
- препараты для коррекции слюнотечения (капают Атропин в рот, делают инъекции ботулотоксина в слюнные железы, применяют гиосциновые пластыри);
-
при психических расстройствах – антидепрессанты (селективные ингибиторы обратного захвата серотонина — Прозак, Серлифт, Золофт) при депрессиях и Вальпроаты при психозах;
- противодиарейные средства: Лоперамид (Имодиум), диетотерапию;
- при развитии инфекционных осложнений со стороны дыхательных путей используют антибиотики, бронхорасширяющие средства (Беродуал), физиотерапевтические процедуры;
- при дистониях и дрожании: антихолинергические препараты (Циклодол, Паркопан, Бипериден, Акинетон).
В последние годы для прекращения накопления сфингомиелина в клетках стали использовать Миглустат. Он блокирует фермент, ответственный за синтезирование гликосфинголипидов (предшественники сфингомиелина).
Применяют в дозировке от 100 мг 1-2 раза в день до 200 мг 3 раза в день (в зависимости от возраста и площади тела больного). Миглустат предотвращает разрушение нервных клеток и, таким образом, замедляет развитие неврологических симптомов, приводит к увеличению продолжительности жизни.
Видимый положительный результат от применения препарата развивается через 6 месяцев- 1 год постоянного приема.

Болезнь Ниманна-Пика
Болезнь Ниманна-Пика (сфингомиелиноз) — группа заболеваний, вызванных генетическими мутациями, влияющими на липидный обмен. Выделяют 3 разновидности:
- Тип A — острая форма заболевания у новорожденных.
- Тип B — умеренная форма. Она реже встречается, носит хронический характер, и ей не присущи неврологические проявления.
- Тип C — биохимически и генетически отдельная форма заболевания.
Иногда выделяют еще тип D. Он был отмечен только у французов, проживающих в небольшом географическом регионе Канады и является разновидностью типа C.
Каждый тип болезни Ниманна-Пика поражает различные органы. В зависимости от вида заболевания может развиваться или отсутствовать поражение нервной системы и дыхательного аппарата. Разные типы этого заболевания отличаются друг от друга симптоматикой.
Подробнее о типах
Тип C значительно отличается от двух других типов заболевания по биохимической и генетической природе. Люди с этим типом болезни не могут метаболизировать холестерин и другие липиды надлежащим образом. В результате избыточное количество холестерина накапливается в печени и селезенке. Подавляющее большинство пациентов с этим типом заболевания умирают в возрасте 20 лет, при этом многие умирают до 10 лет. При более позднем проявлении симптомов отдельные пациенты могут доживать до 40 лет.
Болезнь Ниманна Пика типа A и B встречается во многих этнических группах, но евреи-ашкенази характеризуются повышенной частотой этого заболевания. У этой генетической группы частота болезни составляет 1 на 40000.
При этом один из ста человек является пассивным носителем гена, ответственного за развитие этого заболевания. Сфингомиелиноз типа A и B у других этнических групп встречается примерно в 1 случае на 250000.
Тип A встречается наиболее часто, 85% смертельных исходов от болезни Ниманна — Пика связано именно с этой разновидностью заболевания. Тип C в Западной Европе характеризуется 1 случаем заболевания на 150000 населения.
Причины развития заболевания
Все формы сфингомиелиноза являются наследственными аутосомными рецессивными заболеваниями. Для проявления заболевания требуется присутствие наследственной генетической мутации в одном из генов, ответственных за развитие болезни. Риски развития заболевания равновелики у мужчин и женщин.
Болезнь Ниманна-Пика типа A и B вызывается недостатком определенного фермента (сфингомиелиназы, расщепляющей сфингомиелин).
Этот фермент обычно присутствует в лизосомах (специальных образованиях внутри клеток), он требуется для метаболизма липидов (жиров).
Если этот фермент отсутствует или не функционирует правильно, то это ведет к накоплению жиров в клетке, смерти клеток и сбою в функционировании ряда органов и систем тела.
Сфингомиелиноз типа C является смертельно опасным заболеванием, обусловленным сбоем в хранении жиров. При этом происходит накопление холестерина в печени, селезенке и центральной нервной системе. Мутации в двух независимых генах приводят к клиническим проявлениям этого заболевания.
Какие могут быть симптомы?
Симптомы болезни Ниманна Пика могут значительно варьировать. Диагностика осложняется тем, что и совершенно другие заболевания могут приводить к исходной симптоматике. Ранние стадии заболевания характеризуется проявлением отдельных симптомов, а не всего спектра.
Развитие симптомов сфингомиелиноза типа A начинается в первые месяцы жизни. Среди симптомов присутствуют:
- распухание области живота в течение 3–6 месяцев;
- на глазном дне не появляются красные пятна;
- отсутствие аппетита;
- потеря базовых моторных навыков, усиливающаяся со временем.
Болезнь Ниманна Пика типа B характеризуется более мягкой симптоматикой. Симптомы развиваются в позднем детстве или подростковом периоде.
Увеличение размеров живота может отмечаться у детей младшего возраста. Мозг и нервная система практически не вовлечены в патогенные процессы. У некоторых пациентов могут наблюдаться рецидивирующие инфекции.
Болезнь Ниманна Пика типа C обычно поражает детей школьного возраста. Однако это заболевание может развиться как у грудничков, так и у взрослых людей. Симптомы могут включать:
- уменьшение подвижности конечностей;
- увеличение селезенки;
- увеличение печени;
- желтуха при рождении или вскоре после рождения;
- проблемы с обучаемостью и интеллектуальная деградация;
- эпилептические припадки;
- нарушение речи;
- внезапная потеря тонуса мышц, которая может приводить к падению;
- тремор пальцев рук;
- проблемы с поднятием или опусканием глаз;
- неустойчивая походка, проблемы при ходьбе.
Симптомы сфингомиелиноза типа D, упомянутого выше, сходны с таковыми для типа C.
Методы диагностики
При диагностике сфингомиелиноза типа A и B обычно хватает набора определяющих симптомов. Для облегчения постановки диагноза используется анализ активности сфингомиелиназы в крови. При том, что этот вид анализа эффективно определяет индивидуумов с двумя мутировавшими генами, он не очень надежен для установления случаев, когда мутировал лишь один ген.
Болезнь Ниманна Пика типа C диагностируются посредством биопсии кожи. В лабораторных условиях выращиваются клетки кожи (фибробласты), и анализируется их способность транспортировать и запасать холестерин. Способность клеток транспортировать холестерин оценивается на основании конвертации одной формы холестерина в другую.
Темпы накопления холестерина оцениваются при помощи красящего компонента, флюоресцирующего при ультрафиолетовом освещении. Требуется проведение обоих этих тестов, так как по отдельности они ненадежны.
По причине неверной диагностики сфингомиелиноз типа C зачастую путают с синдромом дефицита внимания, нарушениями обучаемости и замедленным развитием.

- аспирация костного мозга;
- биопсия печени (используется редко);
- анализ глазного дна при помощи щелевой лампы.
Лечение и реабилитация
Обычно требуется привлечение диетолога, физиотерапевта, профпатолога (корректировка моторных навыков, баланс при ходьбе), невролога (лечение эпилептических припадков и оценка неврологического состояния), логопеда, пульмонолога, генетика, гастроэнтеролога, психолога, социального работника и сиделки.

В настоящий момент отсутствует эффективное лечение сфингомиелиноза типа A. Есть определенные положительные результаты в лечении пациентов с типом B при помощи трансплантации костного мозга.
Продолжаются исследования возможных методов лечения, включая генную терапию и заместительную ферментную терапию. Для лечения болезни Ниманна — Пика типа D лекарств не существует на текущий момент.
Препарат под названием миглустат используется для лечения нарушений в нервной системе при типе C.
Больным всеми типами этого заболевания рекомендовано здоровое питание с низким уровнем холестерина. Однако исследования не подтверждают, что такие меры помогут в борьбе с осложнениями или изменят метаболизм холестерина клетками. Однако ряд медицинских препаратов облегчает течение заболевания, например, эпилептические припадки и внезапную потерю мышечного тонуса.
Все виды болезни Ниманна — Пика требуют постоянного ухода и медицинского наблюдения. Продолжительность жизни и ее качество напрямую зависит от типа заболевания и качества предоставляемого медицинского обслуживания.
Сфингомиелиноз типа A относится к крайне тяжелым заболеваниям, обычно он приводит к смерти в возрасте до 2–3 лет. Пациенты с типом B могут доживать до подросткового возраста или взрослого состояния.
Тип C при появлении первых признаков до одного года может приводить к смерти до достижения школьного возраста.
Те пациенты, у которых признаки проявляются после наступления школьного возраста, могут доживать до завершения подросткового периода, а некоторые живут до 20 лет и более.
Возможные осложнения:
- слепота;
- повреждение мозга, сопровождаемое умственной отсталостью и запоздалым развитием двигательных навыков;
- глухота.
В каких случаях следует обращаться к врачам?
К врачам следует обращаться при намерении завести ребенка, и при наличии в семейном анамнезе болезни Ниманна — Пика. Также рекомендуется генетическая консультация и генетический скрининг.
К специалистам следует обращаться при наличии у ребенка симптомов сфингомиелиноза, например:
- проблемы с развитием;
- плохой аппетит;
- малый прирост массы тела.
Меры предосторожности при заболевании
Все типы болезни Ниманна — Пика носят аутосомный рецессивный характер. В результате оба родителя могут являться носителями аномального гена, а внешние признаки заболевания будут отсутствовать.
Если оба родителя являются носителями такого гена, то риск рождения ребенка, больного этим заболеванием, составляет 25%.
При этом риск того, что ребенок будет пассивным носителем этого гена, составляет 50%.
Заболевание проявляется только в том случае, когда ребенок наследует оба рецессивных гена от своих обоих родителей, являющихся скрытыми носителями поврежденного гена.
Для болезни Ниманна — Пика типа A и B ген, ответственный за развитие заболевания, был изолирован и основательно изучен. Это позволяет проводить тестирование ДНК и постановку дородового диагноза. Начиная с начала 1990-х активно ведутся исследования способов лечения этого заболевания.
Текущие разработки фокусируются на трансплантации костного мозга, ферментозаместительной терапии и генной терапии. Все эти виды терапии в лабораторных условиях показали определенную эффективность против сфингомиелиноза типа B.
К сожалению, эти потенциальные виды терапии оказались неэффективны против болезни Ниманна — Пика типа A.
Болезнь Пика

Болезнь Пика — это редкостная, хроническая и прогрессирующая болезнь ЦНС, характеризующаяся атрофией височных, а также лобных долей коры головного мозга с нарастанием слабоумия.
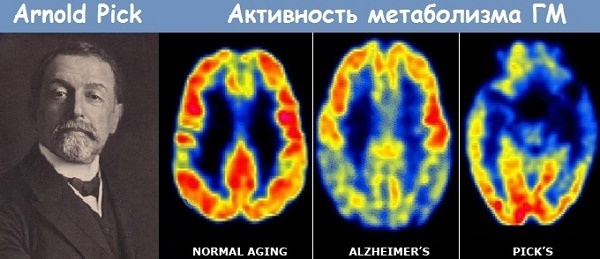
Заболевание начинается в 50-60 лет, хотя бывают и более поздние или ранние манифестации. Женщины склонны болеть чаще мужчин. А. Пик в 1892 году дал описание случаев сенильной деменции, усиливающихся атрофическим процессом главным образом в височных и лобных долях. Подобные исследования проводили А. Альцгеймер, X. Липман, Е. Альтман.
Высказывания о том, что описанные А. Пиком случаи болезни представляют самостоятельную форму, впервые отметил X. Рихтер. Подтверждением этой нозологической самостоятельности заболевания стали проведенные патологоанатомические исследования, показавшие ряд морфологических особенностей именно этой патологии.
Что это такое?
Болезнь Пика — хроническое и прогрессирующее заболевание центральной нервной системы, встречающееся обычно в возрасте 50 — 60 лет и характеризующееся деструкцией и атрофией коры головного мозга преимущественно в области лобных и височных долей. Средний возраст начала заболевания — 54 года, средняя продолжительность до наступления смерти — 6 лет.
Причины развития
Этиология остается невыясненной. Семейные случаи заболевания наталкивают исследователей на мысль о его наследственном характере.
Однако спорадические случаи наблюдаются намного чаще семейных, а в пределах одной семьи чаще болеют братья и сестры, чем родственники в разных поколениях.
Среди возможных этиофакторов названы длительные воздействия на головной мозг вредоносных химических веществ.
Сюда же можно отнести применение наркоза, особенно в случаях его частого использования или неадекватного дозирования.
Некоторые авторы считают, что болезнь Пика может развиться в связи с перенесенным ранее психическим расстройством.
Наряду с этим, исследователи склоняются к мнению, что такие факторы, как интоксикации, черепно-мозговые травмы, инфекции, психические расстройства, гиповитаминоз витаминов гр. В играют лишь провоцирующую роль.
Морфологически определяются атрофические процессы в лобных и височных долях мозга, зачастую больше выраженные в доминантном полушарии. Атрофия затрагивает как кору, так и подкорковые структуры. При этом отсутствуют воспалительные изменения.
Сосудистые нарушения не характерны или слабо выражены. Сенильные бляшки и нейрофибриллярные сплетения, типичные для болезни Альцгеймера, отсутствуют. Патогномоничными для болезни Пика считаются внутринейрональные аргентофильные включения.
Классификация
Различают три стадии развития болезни Пика, каждая из которых характеризуется своей клинической картиной и прогнозом. Следует отметить, что развитие недуга на последней стадии является уже необратимым патологическим процессом и часто провоцирует развитие сопутствующих соматических заболеваний.
Выделяют такие стадии развития недуга:
- первая или начальная — наблюдаются негативные изменения в поведении человека. Чаще всего, это эгоистическая ориентация, раздражённость, агрессия к окружающим;
- вторая — прогрессирование клинической картины первой стадии, ухудшаются интеллектуальные способности человека, отсутствует логическое мышление, больной не может сам справляться с элементарными гигиеническими процедурами;
- глубокое слабоумие, человек нуждается в постоянном уходе.
На последней стадии развития заболевания медикаментозная терапия уже не имеет смысла. В этом случае, основой улучшения качества жизни больного является сестринский уход.
Отличие от болезни Альцгеймера
Болезнь Пика и Альцгеймера имеют один общий признак — слабоумие. Дифференцировать одну болезнь от другой можно по таким критериям:
Симптомы
В начальных стадиях болезни Пика у пациентов наблюдаются такие симптомы: асоциальное поведение, выраженная эгоистичность.
Они утрачивают способность контролировать свои поступки, в результате чего становятся эксцентричными – в частности, удовлетворяют базовые инстинкты, в том числе половое влечение, невзирая на окружающих людей и обстановку.
Критика к своим поступкам снижена. На этом фоне формируются различные расстройства, например булимия или гиперсексуальность.
На начальных стадиях болезни Пика больные могут пребывать как в состоянии эйфории, так и апатии.
У них формируется характерное речевое расстройство, проявляющееся в постоянном повторении одних и тех же анекдотов, фраз, отдельных слов – «симптом граммофонной пластинки».
По мере прогрессирования болезни развивается сенсомоторная афазия – теряется способность словесного выражения мыслей, а также понимание речи окружающих. Затем утрачиваются и другие когнитивные функции: счета (акалькулия), письма (аграфия) и чтения (алексия).
В дальнейшем больные теряют способность последовательно выполнять действия (нарушения праксиса), у них изменяется восприятие окружающего мира (агнозия), происходит потеря памяти (амнезия). Исходом становится глубокая деменция. Пациенты не могут обслуживать себя, дезориентированы во времени и пространстве, обездвижены.
Диагностика
При наличии вышеописанной клинической картины следует обратиться психиатру и неврологу. Диагностика при подозрении на болезнь Пика заключается в следующих мероприятиях:
- на основании физикального осмотра больного и личной беседы, врач оценивает психоэмоциональное состояние пациента;
- КТ и МРТ — для оценки состояния головного мозга;
- электроэнцефалография.
Лабораторные анализы, в этом случае, не представляют какой-либо диагностической ценности. В отдельных случаях может понадобиться биохимический анализ крови.
Следует отметить, что данное заболевание необходимо отличать от следующих:
- рак головного мозга;
- болезнь Альцгеймера;
- хорея Гентингтона;
- психические нарушения при атеросклерозе диффузного типа.
На основании полученных результатов обследования, врач может определить степень развития патологического процесса и выбрать наиболее оптимальную тактику поддерживающей терапии.
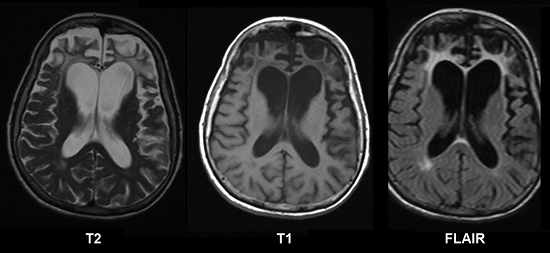
МРТ
Лечение
На сегодняшний день общей методики для лечения болезни Пика не существует. Всё что может сделать врач — назначить медикаментозное лечение для задержки прогрессирующих изменений и улучшения состояния человека.
В лечении болезни Пика применяют ингибиторы холинэстеразы. Это такие препараты, как Амиридин, Ривастигмин (Экселон), Реминил (Галантамин), Арисепт, а также Глиатилин. Эти лекарства при болезни Пика нормализуют состояние пациентов на раннем этапе заболевания.
Хороший эффект имеется от применения длительно (порядка 6 месяцев) блокаторов NMDA (Акатинолмемантин), а также препаратов ноотропного действия (Фенотропил, Аминалон, Ноотропил) и Церебролизин.
Купирование продуктивной психотической симптоматики осуществляется мягкими нейролептиками – Терален, Тералиджен, Клопиксол, Хлорпротиксен.
Пациенты с болезнью Пика нуждаются в постоянной психологической поддержке. Больным рекомендовано участие в специальных тренингах, замедляющих прогрессирование заболевания. Прогноз на будущее неблагоприятный.
Спустя шесть лет после начала заболевания наступает полное моральное, а также психическое разложение личности, наступает маразм и кахексия. Заболевший для общества становится полностью потерян.
Больному нужен обязательный постоянный уход или помещение в специализированную психиатрическую больницу.
Прогноз
Прогноз жизни при деменции, связанной с болезнью Пика неблагоприятный. Болезнь неизлечима, а терапия направлена на ослабление и устранение симптомов.
Так как патология развивается постепенно, то ее наличие долгое время остается незаметным. Пациенты поздно обращаются за медицинской помощью. Это приводит к быстрому прогрессированию болезни и ухудшению состояния пациента.
Живут пациенты с диагнозом Пика от 6 до 10 лет при хорошем уходе и эффективном лечении.
Часто пациенты с деменцией представляют опасность для самих себя и не способны ухаживать за собой – готовить еду, одеваться, мыться, стирать, убирать. Поэтому таким пациентом необходим постоянный уход и контроль.
На третьей стадии патологии пациенты часто становятся обездвиженными вследствие апраксии, дезориентированы. Это приводит к осложнениям – пролежням, инфицированию ран, сепсису, пневмонии.
Осложнения часто приводят к гибели пациента.
При хорошем уходе, эффективном медикаментозном и немедикаментозном лечении, окружении пациента заботой и теплом прогрессирование болезни можно замедлить, частично восстановить когнитивные функции головного мозга и значительно улучшить качество жизни пациента с болезнью Пика.
Болезнь Пика

Болезнь Пика — вариант сенильной деменции с атрофическими изменениями, локализующимися преимущественно в височных и лобных долях мозга. Клинически проявляется нарушением поведения с асоциальными наклонностями и растормаживанием инстинктов, прогрессирующим распадом когнитивных функций. Перечень диагностических мероприятий включает ЭЭГ, УЗДГ церебральных сосудов, Эхо-ЭГ, консультацию психиатра, КТ, СКТ или МРТ головного мозга. Лечение заключается в длительном приеме антихолинэстеразных средств, мемантина, ноотропов, однако оно не позволяет предотвратить полный интеллектуально-мнестический распад личности.
Болезнь Пика представляет собой редкий вид лобно-височной деменции с характерным началом в возрасте 50-60 лет и преимущественной атрофией коры височных и лобных долей мозга. Названа в честь описавшего ее в 1892 г. Арнольда Пика.
Сам Пик считал описанную им патологию лишь отдельным клиническим вариантом старческой деменции.
Однако позже, после выявления подтвержденных паталогоанатомическими исследованиями отличительных морфологических признаков, болезнь Пика была выделена как самостоятельная нозология.
Данные о распространенности заболевания не собраны ввиду его редкости и трудностей прижизненной диагностики. В литературе имеются сведения, что деменция Пика встречается в 4 раза реже болезни Альцгеймера.
Мужчины болеют несколько реже женщин.
Неуклонно и быстро прогрессирующий характер патологии, трудности ее диагностирования и терапии делают болезнь Пика актуальной проблемой современной геронтологии и неврологии.
Болезнь Пика
Этиология остается невыясненной. Семейные случаи заболевания наталкивают исследователей на мысль о его наследственном характере.
Однако спорадические случаи наблюдаются намного чаще семейных, а в пределах одной семьи чаще болеют братья и сестры, чем родственники в разных поколениях. Среди возможных этиофакторов названы длительные воздействия на головной мозг вредоносных химических веществ.
Сюда же можно отнести применение наркоза, особенно в случаях его частого использования или неадекватного дозирования. Некоторые авторы считают, что болезнь Пика может развиться в связи с перенесенным ранее психическим расстройством.
Наряду с этим, исследователи склоняются к мнению, что такие факторы, как интоксикации, черепно-мозговые травмы, инфекции, психические расстройства, гиповитаминоз витаминов гр. В играют лишь провоцирующую роль.
Морфологически определяются атрофические процессы в лобных и височных долях мозга, зачастую больше выраженные в доминантном полушарии. Атрофия затрагивает как кору, так и подкорковые структуры. При этом отсутствуют воспалительные изменения.
Сосудистые нарушения не характерны или слабо выражены. Сенильные бляшки и нейрофибриллярные сплетения, типичные для болезни Альцгеймера, отсутствуют. Патогномоничными для болезни Пика считаются внутринейрональные аргентофильные включения.
В своем развитии болезнь Пика проходит 3 последовательные стадии. В начальной стадии преобладают изменения личности с утратой выработанных моральных принципов. Человек становится крайне эгоистичен, наблюдается асоциальное поведение.
Расторможенность инстинктов и потеря контроля над своими поступками приводят к немедленной реализации инстинкта (физиологических потребностей, полового влечения), независимо от окружения и обстановки.
Критика пациента значительно снижена и он объясняет свое поведение тем, что «не смог удержаться», «так получилось», «не терпелось». На этом фоне возможно развитие булимии, гиперсексуальности и др. расстройств.
Изменения речи сводятся к многократным повторам слов, фраз, шуток, историй или секретов (симптом «граммофонной пластинки»). Отмечается эйфория или апатия.
Преобладание в клинике начального периода деменции Пика тех или иных проявлений зависит от локализации появляющихся атрофических процессов в головном мозге.
Так, базальная фронтальная атрофия сопровождается личностными расстройствами, эмоциональной неустойчивостью, сменой периодов ригидности и расторможенности; конвекситальная атрофия лобной доли — сочетанием асоциального поведения с апатией и абулией — утратой побуждений и желаний, полным безволием.
При преобладании атрофии в левом полушарии нарушения поведения проявляются на фоне депрессии. Правополушарная атрофия характеризуется неадекватным поведением в сочетании с эйфорией.
Во второй стадии появляются и нарастают когнитивные нарушения. Возникает сенсомоторная афазия — пациент не может сформулировать свои мысли и утрачивает способность понимать речь окружающих. Наблюдаются алексия, аграфия и акалькулия — потеря навыков чтения, письма и счета.
Отмечаются амнезия — потеря памяти, агнозия — изменение восприятия мира и нарушения праксиса — способности последовательно выполнять действия. Вначале расстройство когнитивных функций может носить эпизодический характер, затем оно становится постоянным и прогрессирует вплоть до полного интеллектуального разрушения личности.
У ряда пациентов своеобразным проявлением становится кожная гиперальгезия (повышенная чувствительность).
Третья стадия болезни Пика — глубокая деменция. Пациенты обездвижены вследствие крайней апраксии, дезориентированы, не способны к элементарным действиям по самообслуживанию, нуждаются в постоянной опеке.
Такое состояние приводит к гибели больных, обусловленной церебральной недостаточностью или интеркуррентными инфекциями (инфицированием пролежней с развитием сепсиса, застойной пневмонией, восходящим пиелонефритом и т. п.).
Немаловажное значение в диагностике имеет опрос пациента и его близких, исследование анамнеза, объективный осмотр. Пирамидная симптоматика в неврологическом статусе не характерна.
При вовлечении в атрофический процесс подкорки возможно выявление признаков поражения экстрапирамидной системы (гиперкинезов, проявлений вторичного паркинсонизма). Соматическое состояние в 1-2 стадии деменции Пика, как правило, удовлетворительное.
В ходе диагностики следует дифференцировать болезнь Пика от сосудистой деменции, болезни Альцгеймера, внутримозговых опухолей с локализацией в лобных долях, артериовенозной мальформации головного мозга, шизофрении.
Клинические данные (возраст начала болезни, ее манифестация с поведенческих нарушений с последующим присоединением когнитивной дисфункции, отсутствие общемозговой и пирамидной симптоматики и пр.
) позволяют неврологу предположить болезнь Пика лишь во второй стадии, когда имеются когнитивные нарушения.
Изменения в поведении, характеризующие дебют заболевания, зачастую принимаются за психические расстройства, в связи с чем родственники больного в первую очередь обращаются за помощью к психиатру.
При проведении ЭЭГ у пациентов с деменцией Пика выявляется снижение вольтажа биоэлектрической активности в лобных отведениях. РЭГ и транскраниальная УЗДГ не выявляют существенных сосудистых нарушений. По данным Эхо-ЭГ может диагностироваться умеренная гидроцефалия.
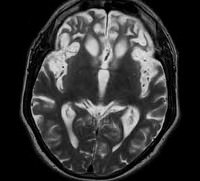
Визуализировать атрофические изменения мозга позволяют томографические методы: КТ, МСКТ и МРТ.
На МРТ головного мозга выявляется истончение коры, участки атрофического уменьшения плотности мозгового вещества в лобных и височных долях, расширение субарахноидальных пространств.
Специфическая терапия не разработана. В схему лечение, как правило, включают антихолинэстеразные фармпрепараты (холина альфосцерат, галантамин, ривастигмин, донепезил), ноотропы (пирацетам, фонтурацетам, гамма-аминомасляная кислота), мемантин.
При наличии агрессивного поведения назначают нейролептики: алимемазин, хлорпротиксен.
В начальных стадиях рекомендована психотерапия, участие в когнитивных тренингах, консультирование психолога; в более поздних — сенсорная комната, арт-терапия, симуляция присутствия.
Все вышеуказанные методы направлены на замедление прогрессирования симптомов. Однако их эффективность при деменции Пика существенно ниже, чем при болезни Альцгеймера. Несмотря на проводимую терапию, состояние глубокой деменции наступает в среднем через 5-6 лет после дебюта болезни. Продолжительность жизни от начала заболевания не превышает 10 лет.
Comments
(0 Comments)