Болезнь Бурневилля
Патогенез
Основные звенья патогенеза:
- Появление в крови антител к собственным клеткам организма,
- Токсичное влияние Т-лимфоцитов на эпителиоциты слизистой,
- Отсутствие IgA в слюне,
- Формирование язв и очагов некроза на слизистой,
- Повышение количества фагоцитов в крови,
- Повреждение эндотелия кровеносных сосудов.
В результате перечисленных выше изменений, происходящих в организме больного, воспаляются стенки артерий и вен, на эндотелии в периваскулярном пространстве образуются лимфоцитарные и плазмоцитарные инфильтраты, утолщается внутренняя оболочка сосудов, сужается их просвет.
Аутосомная рецессивная агаммаглобулинемия швейцарского типа
Тяжелый комбинированный иммунодефицит (SCID, ТКИД), или аутосомная рецессивная агаммаглобулинемия швейцарского типа, является наиболее серьезным расстройством из группы человеческих иммунодефицитов. В медицинских источниках и литературе данное заболевание известно также под названием алимфоцитоз. Это заболевание врожденного характера.
Развитие ТКИД сопровождается нарушениям работы гуморальной части иммунной системы человека. Клетки, которые должны производить иммунный ответ, не функционируют. Дети с данным заболеванием подвержены повторяющимся тяжелым инфекциям, задержке роста и ранней смерти.
ТКИД встречается у одного из пятисот тысяч новорожденных. Понятие «тяжелый комбинированный иммунодефицит» объединяет несколько заболеваний, в частности:
- агаммаглобулинемия швейцарского типа;
- дефицит аденозиндезаминазы;
- аутосомно-рецессивный тяжелый комбинированный иммунодефицит;
- синдром голых лимфоцитов;
- ТКИД с лейкопенией. У детей с этой формой заболевания отсутствуют белые клетки крови — гранулоциты.
Для того чтобы понять, почему ТКИД считается самым тяжелым расстройством среди всех типов иммунодефицитов, можно рассмотреть схему иммунной системы человека. Она состоит из трех частей: клеточной, гуморальной и неспецифической. Клеточные и гуморальные части системы необходимы для борьбы с инфекциями — они определяют возбудителей болезней и атакуют их. Клеточная система состоит из множества классов Т-лимфоцитов (белых кровяных клеток, которые обнаруживают инородных захватчиков – антигены). Гуморальная система состоит из В-клеток, которые являются единственными клетками в организме, производящими антитела. При ТКИД ни клеточная, ни гуморальная часть иммунной системы не функционирует так, как необходимо.
Причины и симптомы
SCID является наследственным заболеванием. Существует два типа неправильного развития иммунной системы плода. При первом типе генетического нарушения В- и Т-клетки являются дефектными. При втором типе только Т-клетки являются аномальными, но их недостаток влияет на функционирование лимфоцитов.
В течение первых нескольких месяцев жизни ребенок с ТКИД защищен антителами, попавшими в его организм из крови матери. Однако уже в возрасте трех месяцев ребенок с подобным нарушением начинает страдать от грибковой инфекции полости рта (молочницы), хронической диареи, отита и легочных инфекций, в том числе пневмоцистной пневмонии. Ребенок теряет вес, становится очень слабым, и, в конце концов, умирает от условно-патогенной инфекции.
Лечение аутосомной рецессивной агаммаглобулинемии
Тяжелый комбинированный иммунодефицит/агаммаглобулинемию можно лечить с помощью антибиотиков и иммунной сыворотки. Однако такие методы лечения не могут защитить больных, а лишь защищают от инфекций. Постоянная защита невозможна из-за привыкания к антибиотикам и уничтожения полезной микрофлоры ЖКТ. В настоящее время один из немногих эффективных методов лечения этого заболевания — это пересадка костного мозга.
Клинические проявления
Первые симптомы заболевания проявляются, как правило, в возрасте менее 1 года, чаще всего после 3-4 месяцев жизни. Это связано с постепенным снижением количества антител, полученных от матери. Больные страдают рецидивирующими инфекциями, вызываемыми , и другими пиогенными бактериями. против может осложняться полиомиелитом. вызывает прогрессирующий часто фатальный вирусный . Инфекция или ведёт к хронической и . Первично поражаются , придаточные пазухи носа. В клинической картине отмечается , синдром мальабсорбции, , поражения ЦНС (энцефалиты), , . Возможны системные ревматические проявления по типу диффузных болезней соединительной ткани. Суставной синдром характеризуется эпизодической мигрирующей полиартралгией либо крупных . Даже при длительном течении артрит не приводит к рентгенологическим изменениям пораженных суставов. Имеют место кожные поражения — , .
Диагностика
При появлении лихорадки и общей слабости, резком похудении необходимо обратиться к врачу-ревматологу.
Диагностика болезни Бехчета, как и всех остальных васкулитов, очень сложна. Проводят ее в специализированных медицинских учреждениях с помощью микроскопических, иммунологических, химических и инструментальных методов исследования. Недопустимо самостоятельно диагностировать васкулит и проводить самолечение.
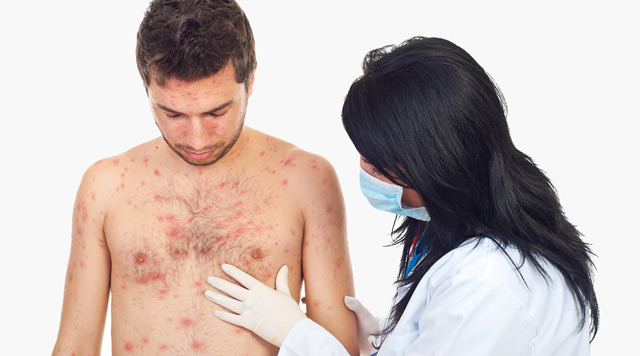
Диагностика болезни Бехчета основывается на результатах клинико-диагностического обследования больных. Патология имеет рецидивирующее течение, проявляется симптомами стоматита, поражения гениталий, воспаления суставов, сосудистой оболочки глаза, головного мозга. Диагностикой и лечением патологии кроме ревматологов занимаются врачи-неврологи, дерматологи и прочие специалисты.
В общем анализе крови обнаруживают анемию, лейкоцитоз со сдвигом формулы влево, повышение СОЭ. Диагностика артрита заключается в изучении данных, полученных на рентгенографии, УЗИ суставов и артроскопии. Пункция сустава и биопсия пораженных тканей с их последующим морфологическим изучением позволяют подтвердить или опровергнуть предполагаемый диагноз. Инструментальные методы, выявляющие изменения в суставах и неврологические нарушения: рентгенография грудной клетки, УЗИ органов брюшной полости, МРТ.
Литература
- Иммунология детского возраста: практическое руководство по детским болезням под ред. А. Ю. Щербины и Е. Д. Пашанова. М.: Медпрактика-М, 2006.
- Ballow M. Primary immunodeficiency disorders: antibody deficiency // J Allergy Clin Immunol 2002; 109:581-91
- Педиатрия, Шабалов Н. П., СПб.: Спецлит, 2005.
- Педиатрия, Дж. Греф, М.: Практика, 1997.
X-сцепленное рецессивное наследование (англ. X-linked recessive inheritance) — один из видов сцепленного с полом наследования. Такое наследование характерно для признаков, гены которых расположены на Х-хромосоме и которые проявляются только в гомозиготном или гемизиготном состоянии. Такой тип наследования имеет ряд врождённых наследственных заболеваний у человека, эти заболевания связаны с дефектом какого-либо из генов, расположенных на половой Х-хромосоме, и проявляются в случае, если нет другой Х-хромосомы с нормальной копией того же гена. В литературе встречается сокращение XR для обозначения X-сцепленного рецессивного наследования.
Для Х-сцепленных рецессивных заболеваний характерно, что обычно поражёнными являются мужчины, для редких Х-сцепленных заболеваний это справедливо почти всегда. Все их фенотипически здоровые дочери являются гетерозиготными носительницами. Среди сыновей гетерозиготных матерей соотношение больных и здоровых равно 1 к 1.
Частным случаем Х-сцепленного рецессивного наследования является крисс-кросс наследование (англ. criss-cross inheritance, также наследование крест-накрест), в результате которого признаки отцов проявляются у дочерей, а признаки матерей — у сыновей. Название такому типу наследования дал один из авторов хромосомной теории наследования Томас Хант Морган. Он впервые описал такой тип наследования для признака цвета глаз у дрозофилы в 1911 году. Крисс-кросс наследование наблюдается тогда, когда мать является гомозиготой по рецессивному признаку, локализованному в Х-хромосоме, а у отца в единственной Х-хромосоме имеется доминантный аллель этого гена. Выявление такого вида наследования при анализе расщепления является одним из доказательств локализации соответствующего гена на Х-хромосоме.
Иммунодефициты — нарушения иммунологической реактивности, обусловленные выпадением одного или нескольких компонентов иммунного аппарата или тесно взаимодействующих с ним неспецифических факторов.
Единой классификации не существует.
По происхождению иммунодефициты делят на первичные и вторичные.
Причины, симптомы и методы лечения гипогаммаглобулинемии
В организме существует два вида лимфоцитов. Т-лимфоциты отвечают за уничтожение болезнетворных организмов, В-лимфоциты производят антитела, «налипающие» на атакующие организм патогены, таким образом, Т-лимфоциты и другие защитные клетки накапливаются и атакуют чужеродные организмы.
Антитела – это ключевые белки, ответственные за обнаружение болезнетворных патогенов, понижение их уровня увеличивает восприимчивость к инфекциям. Критическое снижение количества антител вызывает гипогаммаглобулинемию.
Причины
Гипогаммаглобулинемия может быть первичной (то есть, врожденной) или вторичной (приобретенной), вызванной заболеваниями. Первичная форма заболевания может проявиться как при рождении, так и позднее — в более старшем возрасте, несмотря на то, что дефектный ген в организме присутствует с рождения. Большинство случаев гипогаммаглобулинемии развивается после 6-месячного возраста. Случаи заболевания, проявляющиеся на 3-м или 4-м десятилетии жизни, принято считать первичным иммунодефицитом. Их развитие, полагают медики, является результатом еще не установленных генных дефектов.
Симптомы
Большинство пациентов с гипогаммаглобулинемией страдают от рецидивирующих инфекций. Острота проявления симптомов зависит от семейной истории болезни, возраста начала болезни, имеющихся в организме инфекций, состава крови, состояния ЖКТ и опорного аппарата, а также от того, имеются ли у пациента аутоиммунные заболевания или болезни сосудов.
Симптомы гипогаммаглобулинемии:
- задержка роста;
- аномалии лимфоидной ткани и органов (например, нехватка тканей миндалин, аденоиды, малый размер периферических лимфатических узлов);
- аномалии развития (например, скелета или грудной клетки);
- дефекты кожи и слизистых оболочек (рубцы, сыпь или сетчатое ливедо);
- ушные, носовые, горловые дефекты (например, перфорация барабанной перепонки, гнойные выделения из носа, сетчатая структура слизистой оболочки глотки);
- легочные нарушения (бронхоэктазы и легочный фиброз с хрипами, одышка и другие);
- сердечно-сосудистые нарушения (громкий шум в легочной артерии сердца, недостаточность трехстворчатого клапана, шум, предполагающий легочную гипертензию);
- гепатомегалия;
- отек нижних конечностей;
- неврологические нарушения (паралитический полиомиелит или глубокая потеря чувствительности со снижением вибрационных воздействий и снижением чувствительности конечностей).
Лечение
Лечение гипогаммаглобулинемии направлено на устранение основной причины недомогания, заболевание или инфекцию. Пациентам с нарушением функции Т-клеток, тяжелыми расстройствами В-клеток не назначают живые вакцины. Высокие дозы ВВИГ (внутривенного иммуноглобулина) или интератекального иммуноглобулина могут быть полезны пациентам с энтеровирусным менингоэнцефалитом. Показано применение полиэтиленгликоля при дефиците аденозиндезаминазы. Для лечения гранулематозных заболеваний применяют фактор некроза опухолей (ФНО). Успешным методом лечения является и генная терапия, наиболее эффективна она у детей с агаммаглобулинемией Брутона. У взрослых и детей старшего возраста эффективность такого метода лечения ниже.
Комментарии
(0 Комментариев)