Фенилкетонурия: 3 метода диагностики, основные симптомы и 4 важных правила диетотерапии
1.4.3. Фенилкетонурия

(классификация
дана по Mc
Kusik
V.A.,
1988)
ФЕНИЛКЕТОНУРИЯ
1.
Классическая
фенилкетонурия (ФКУ) описана А.
Folling.,1934
г.
Заболевание
наследуется
аутосомно-рецессивно
и вызвано мутацией гена, локализующегося
в длинном плече 12 хромосомы.
В
основе
болезни
лежит дефицит фермента фенилаланин -4-
гидроксилазы, обеспечивающего превращение
фенилаланина в тирозин. В результате
метаболического блока происходит
значительное накопление в тканях и
жидкостях больного организма фенилаланина
и таких его производных, как
фенилпировиноградная, фенилмолочная,
фенилуксусная кислоты, фенилэтиламин,
фенилацетилглютамин,
и др.
В
патогенезе ФКУ
имеют значение следующие механизмы:
Прямое токсическое действие на ЦНС фенилаланина и его производных;
Нарушение в обмене белков, липо- и гликопротеидов;
Расстройства транспорта аминокислот;
Нарушение метаболизма гормонов;
Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина);
Нарушение функции печени : диспротеинемия, генерализованная гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение окислительной и белковосинтезирующей функции клеточных органелл.
Частота
классической
ФКУ среди новорожденных по данным
массового скрининга в среднем колеблется
от 1 : 5000 до 1 : 10000 по разным регионам
России.
ФЕНИЛКЕТОНУРИЯ
2.
Впервые
атипичная ФКУ описана I.
Smith,
1974 г.
Заболевание связано с дефицитом
дигидроптеридинредуктазы.
Заболевание
наследуется аутосомно-рецессивно.
Генный дефект локализуется в коротком
плече 4 хромосомы, участке 4р 15.3.
Обратите внимание
В
результате недостаточности
дигидроптеридинредуктазы нарушается
восстановление активной формы
тетрагидробиоптерина, участвующего в
качестве кофактора в гидроксилировании
фенилаланина, тирозина, и триптофана.
Частота
заболевания
составляет 1 : 100 000 новорожденных.
Рано
начатое лечение способствует нормализации
фенилаланина в крови, однако не
предупреждает появление клинической
симптоматики, которая развивается в
начале второго полугодия жизни.
Фенилкетонурию 2 называют диеторезистентной
ФКУ.
ФЕНИЛКЕТОНУРИЯ
3.
Этот
вариант болезни описал S.
Kaufman
в
1978 г. Заболевание наследуется
аутосомно-рецессивно и связано с
недостаточностью 6-пирувоилтетрагидроптерин
синтетазы, участвующей в процессе
синтеза тетрагидробиоптерина.
Развивающиеся при этом расстройства
сходны с нарушениями, наблюдаемыми при
ФКУ 2.
Частота
болезни
составляет 1 : 30 000 новорожденных.
Фенилкетонурия
3 также диеторезистентна
ДРУГИЕ
ВАРИАНТЫ ФКУ.
Эти
формы ФКУ связаны с нарушением
альтернативных путей обмена фенилаланина.
Формируется метилминдальная
ацидурия и парагидроскифенилуксусная
ацидурия.
МАТЕРИНСКАЯ
ФЕНИЛКЕТОНУРИЯ.
Заболевание
развивается у потомков женщин, страдающих
ФКУ и не получающих диету в зрелом
возрасте. Патогенез
мало
изучен, предполагается, что он сходен
с патогенезом остальных форм ФКУ.
Тяжесть
поражения плода коррелирует с уровнем
фенилаланина в плазме матери. Так
как эмбрион особенно чувствителен
к тератогенным воздействиям , рекомендуется
начинать диету еще до наступления
беременности.
В суточном рационе
использовать менее 15-20 мг/кг
фенилаланина. При это важно избегать
дефицита незаменимых аминокислот.
КЛИНИКА
ФЕНИЛКЕТОНУРИИ.
При
рождении больные фенилкетонурией не
отличаются от других новорожденных.
Манифестация ФКУ происходит обычно в
возрасте 2-6 месяцев.
Первыми
проявлениями болезни
служат:
- вялость ребенка, отсутствие интереса к окружающему;
- повышенная раздражительность, беспокойство;
- срыгивание, рвота;
- судорожные эквиваленты: спонтанный рефлекс Моро, спонтанный рефлекс Бабинского, сосательные автоматизмы, приапизм у мальчиков, атетозные движения;
- судорожный синдром;
- заплесневелый, мышиный, волчий запах мочи и пота.
При
отсутствии лечения формируется задержка
статико-моторного и психоречевого
развития, умственная отсталость
достигает, как правило, глубокой
степени.
Характерны
такие фенотипические особенности, как
экзематозные и себорейные сыпи,
гипопигментация кожи, волос, радужной
оболочки глаз.
ДИАГНОСТИКА
ФЕНИЛКЕТОНУРИИ.
ФКУ
может быть диагностирована на основе
обнаружения следующих признаков:
- стойкой гиперфенилаланинемии (более 240 ммоль/л);
- вторичного дефицита тирозина;
- экскреции фенилкетонов с мочой (проба Феллинга на экскрецию фенилпировиноградной кислоты).
В
настоящее время, согласно приказу
Минздрава России № 316 от 30.12.93 проведение
неонатального скрининга на ФКУ стало
обязательным. Скрининирующие тесты
должны быть простыми, недорогими и
информативными. Этим требованиям
отвечают методы, используемые для
ранней диагностики ФКУ:
- микробиологический тест Гатри;
- метод флюоресцирующих антител (лабораторный комплекс “Флюороскан”, позволяющий проводить 800 проб в час);
- метод тонкослойной хроматографии .
Оптимальные
сроки обследования новорожденных —
доношенных, зрелых — 5-6 день жизни;
недоношенных, незрелых, больных —
10-14 день жизни.
Трактовка результатов:
1 группа
— уровень фенилаланина не превышает 200
ммоль/л
(1-3 мг%)
— норма;
2 группа
— уровень фенилаланина составляет
200-500
ммоль/л
(3-10 мг%) — гиперфенилаланинемия. В
эту группу входят дети с транзиторной
гиперфенилаланинемией, вследствие
незрелости ферментных систем печени и
больные ФКУ.
За данной группой проводится
наблюдение по следующему плану: если в
течение 6 недель при еженедельном
исследовании уровень фенилаланина
остается менее 500 ммоль/л, контроль за
уровнем фенилаланина крови проводят
до 1 года первоначально каждые 3 месяца,
а затем каждые 6 мес. жизни.
При уровне
более 500 ммоль/л назначается диетотерапия.
Важно
3 группа
— уровень фенилаланина превышает 500
ммоль/л
(более 10 мг%), диагностируется ФКУ и с
момента постановки диагноза назначается
диетотерапия.
В
настоящее время разрабатываются и
внедряются молекулярно-генетические
методы диагностики генного дефекта при
ФКУ. Прямая диагностика мутантного гена
проводится с помощью синтетических
олигонуклеотидных зондов, этот метод
пригоден для дородовой диагностики ФКУ
и выявления гетерозиготного носительства.
Помимо
молекулярно-генетического анализа,
выявление гетерозигот может осуществляться
биохимическими тестами после нагрузки
фенилаланином в дозе 25 мг/кг.
Диеторезистентные
формы ФКУ диагностируют при помощи:
- исследования биоптеринов мочи ;
- перорального нагрузочного теста с тетрагидробоиптерином (через 4-6 часов после однократной дачи нагрузки в дозе 7,5 мг/кг массы тела происходит резкое снижение и нормализация уровня фенилаланина в крови с одновременным повышением уровня тирозина);
- исследования активности дигидроптеридинредуктазы и 6-пирувоилтетрагидроптерин синтетазы в культуре кожных фибробластов, эритроцитах, гепатоцитах.
ЛЕЧЕНИЕ
ФЕНИЛКЕТОНУРИИ.
Главным способом лечения является диетотерапия, ограничивающая поступление в организм фенилаланина; приступить к ней нужно немедленно после установления диагноза.
Из
рациона больных исключаются:
молоко,
молочные продукты, творог, мясо,
мясные продукты, колбасы, рыба, яйца,
хлебобулочные изделия, фасоль, горох,
орехи, шоколад.
Белковым
эквивалентом этих продуктов питания
становятся гидролизаты белка, либо
аминокислотные смеси, лишенные
фенилаланина: “Лофенолак”, “Фенилфри”
(США), “Берлофен”, “Апонти”, “Гипофенат”
у детей до 4-5 лет и “Нофелан” — у детей
старше 5 лет.
В
пищевой рацион входят овощи, фрукты,
мед, растительное масло, безбелковый
хлеб.
Наиболее
рационально отменять
диетическое лечение
в
возрасте 7-8
лет.
Препараты с промедиаторным действием:
- Наком (комбинация карби ДОФА и лево ДОФА) — доза 100-375 мг/сутки в течение 3-4 недель, перерыв между курсами 1,5-2 месяца.
- Лево-дофа — доза 10-15 мг/кг в сутки;
- 5-окситриптофан — доза 10 мг/кг сутки.
Диеторезистентные формы. Лечение включает назначение тетрагидробиоптерина — доза 10-20 мг/кг в сутки.
ПРОФИЛАКТИКА
ФЕНИЛКЕТОНУРИИ.
- Выявление гетерозиготных носителей;
- Внедрение программ массового скрининга новорожденных для раннего выявления ФКУ и своевременного назначения диетотерапии;
- Пренатальная диагностика.
Фенилкетонурия: основные сведения
Фенилкетонурия – довольно редкое заболевание. Согласно статистике, 1 и 10 000 появившихся на свет новорожденных не обладает способностью усваивать фенилаланин – одну из самых распространенных в рационе питания человека аминокислот.
Коварство этой болезни заключается в том, что без проведения специального анализа крови выявить ее наличие не представляется возможным, а обычная пища превращается в натуральный яд, отравляющий день за днем, год за годом мозг маленького человека.
ФКУ без лечения приводит к самым тяжелейшим последствиям, проявляющимся в постепенном развитии слабоумия, вплоть до олигофрении и идиотии.
Однако не все так уж и безнадежно, терапия фенилкетонурии существует и заключается в соблюдении специальной лечебной диеты в течение всей жизни.
Симптомы ФКУ
Первые симптомы фенилкетонурии можно заметить у малыша уже в трехмесячном возрасте. Чаще всего медики и родители обращают внимание на:
- Отставание в детском развитии
- Эпилептические приступы
- Нарушения мышечной спастики
- Слишком маленький размер мозга
- Неприятный запах мочи и пота (ацетон)
- Недостаток пигмента (обычно больные отличаются светлым цветом кожи и волос, голубыми глазами).
Диагностика ФКУ не представляет особой сложности. Для ее проведения достаточно лишь капли крови, взятой из пятки малыша через пару дней после рождения. В настоящее время анализ крови на фенилкетонурию входит в список обязательных процедур скрининга всех новорожденных.
Лечебная диета
Так как большинство продуктов правильного питания обычного человека содержит в своем составе фенилаланин (мясо, рыба, яйца, злаковые, молоко и кисломолочные продукты), ребенку, которому поставлен диагноз фенилкетонурия, требуется обеспечить соблюдение специальной безбелковой диеты. Аминокислоты, необходимые организму для роста и регенерации тканей, должны поступать в виде искусственно производимых смесей. Придерживаться диеты необходимо не только до полового созревания, но зачастую и пожизненно.
Кроме этого больным необходимо производить постоянный контроль уровня фенилаланина в крови.
Профилактика фенилкетонурии
Так как ФКУ является врожденным заболеванием генетического характера, никаких мер профилактики этой болезни не существует. Если семейный анамнез позволяет предполагать возникновение фенилкетонурии у будущего ребенка, женщине уже во время беременности стоит перейти на специальную диету, чтобы исключить вероятность негативного влияния фенилаланина на развивающийся мозг растущего плода.
Фенилкетонурия

Фенилкетонурия — врожденное аутосомно-рецессивное нарушение с мутацией в хромосоме 12. Отсутствие активности фенилаланингидроксилазы в печени приводит к накоплению фенилаланина и его метаболитов в крови, моче, ликворе; при этом тирозин и катехоламины в дефиците.
Фенилкетонурия всегда сопровождается умственной отсталостью. В США 1 : 50 белых является носителем и 1:12000 страдает фенилкетонурией.
В норме содержание фенилаланина в крови 2 мг/дл.
Результаты анализов при фенилкетонурии
- Классическая форма фенилкетонурии: высокий уровень фенилаланина в крови (обычно более 30 мг/дл, в раннем детстве более 20 мг/дл), повышено содержание фенилаланина и его метаболитов в моче; уровень тирозина в норме или ниже нормы.
- Среднетяжелые формы фенилкетонурии: уровень фенилаланина в крови 15-30 мг/дл и в моче всегда присутствуют метаболиты (частота присутствия 1 : 15000).
- Легкая персистирующая гиперфенилаланинемия: уровень фенилаланина в крови 2-12 мг/дл и метаболитов в моче может не быть (частота 1 : 30000).
Для скрининга новорожденных на фенилкетонурию при уровне фенилаланина в крови менее 15 мг/дл количество фенилпируватной кислоты в моче может быть недостаточным для подтверждения диагноза колориметрическим методом. Кроме того, фенилпирувата может не быть в моче первые 2-3 недели жизни.
Ложно-отрицательный результат может быть получен в тесте Гатри со штаммами микроорганизмов, если кровь собирать в капиллярную трубку, а не на фильтровальную бумагу. Предварительные тесты позволяют определить только уровень фенилаланина более 4 мг/дл. Диагностика фенилкетонурии достовернее при нагрузочном тесте.
Если повторные тесты положительные, то проводят количественный тест уровня фенилаланина и тирозина в крови для подтверждения фенилаланинемии и исключения преходящей формы тирозинемии, которая часто встречается у новорожденных с положительными предварительными тестами. Для этого используют масс-спектрометрию и флуорометрию.
Совет
Серийные обследования должны быть проведены и у пациентов с нелеченными пограничными состояниями, потому что впоследствии в результате стресса или инфекции болезнь может проявиться.
Диагноз фенилкетонурии дополнительно подтверждают оценкой уровня метаболитов в крови и моче после нагрузочного теста: прием 100 мг аскорбиновой кислоты и забор материала спустя 24 часа.
Успешность диетотерапии при фенилкетонурии контролируется в течение жизни по уровню фенилаланина в крови:
- До 12 лет уровень фенилаланина 2-6 мг/дл.
- После 12 лет: 2-10 мг/дл.
- В юности: 2-15 мг/дл.
В течение беременности норма фенилаланина 2-5 мг/дл, потому что с повышением уровня фенилаланина в сыворотке возрастает риск микроцефалии, врожденных аномалий сердца, умственной отсталости.
Рекомендации по частоте контроля за уровнем фенилаланина в крови:
- 1-й год жизни: 1 раз в неделю.
- с 1 года до 12 лет: 2 раза в месяц.
- после 12 лет: 1 раз в месяц.
- беременным с фенилкетонурией: 2 раза в неделю.
В процессе лечения уровень фенилаланина в крови необходимо контролировать (2 раза в неделю первые 6 месяцев, 1 раз в неделю следующие 6 месяцев, 2 раза в месяц до 18 месяцев жизни, 1 раз в месяц в дальнейшем).
Диету выверяют, контролируя уровень фенилаланина в крови (например, 10 мг/дл с последовательно отрицательным тестом в моче). У беременных с нелеченной фенилкетонурией и высоким уровнем фенилаланина в крови резко возрастает частота развития у плода умственной отсталости, врожденных пороков сердца, микроцефалии.
Поиск гетерозиготных носителей и предродовая диагностика возможны с помощью анализа ДНК.
75% гетерозигот выявляется при анализом ДНК в указанные периоды, а также при пренатальной диагностике.
У 15% пациентов с фенилкетонурией выявляются врожденные пороки сердца.
Для диагностики фенилкетонурии применяется анализ с полосками хлорида железа, под действием фенилпирувата мочи они окрашиваются.
Субстанция
Фенилкетонурия
Преходящая тирозинемия
Фенилаланин сыворотки
более 15 мг/дл
более 4 мг/дл (15-20 мг/дл)
Тирозин сыворотки
менее 5 мг/дл (выше не бывает)
более 4 мг/дл (5-20 мг/дл)
Гидроксифенилуксусная кислота в моче
присутствует
отсутствует
Моча
фенилаланин более 100 мкг/мл
широкие колебания тирозина и его метаболитов
Фенилкетонурия

В зависимости от недостатка фермента, который участвует в процессе превращения фенилаланина в тирозин, выделяются следующие формы (каждый из указанных ферментов берет участие в переходе тирозина в фенилаланин, но на различных этапах):
- классическая либо тяжелая (недостаток фенилаланин-4-гидроксилазы);
- атипичные формы (они устойчивы относительно диетотерапии, потому лечить их гораздо сложнее, так как именно диета – основной способ излечения данного заболевания):
- недостаток дегидроптеринредуктазы;
- недостаток пирувоилтетрагидроптеринсинтазы.
Фенилкетонурия у детей – это генетический дефект, который передается по наследству.
Причины фенилкетонурии:
- браки среди близких родственников, которые повышают вероятность развития болезни у новорожденного ребенка;
- изменение гена (мутация) по различным причинам.
При такой болезни, как фенилкетонурия, симптомы проявляются уже на первых неделях или месяцах жизни малыша, но только в том случае, если фенилкетонурия не была обнаружена вовремя. Признаки фенилкетонурии:
- альбинизм: очень светлая кожа и голубой цвет глаз;
- своеобразный запах от кожи малыша, похожий на мышиный;
- высыпания аллергического характера на коже: небольшие бугорки (папулы), маленькие пузырьки с прозрачным содержимым (везикулы), покраснения на коже;
- слабость и вялость;
- судороги.
- медленное умственное и физическое развитие;
- нарушения в умственном развитии, от самых легких форм до идиотии, развиваются в более позднем возрасте.
Диагностика фенилкетонурии проводится в родильном доме на пятый день рождения малыша. С этой целью после кормления берется кровь из пятки ребенка и исследуется уровень фенилаланина. Также возможно исследование мочи. Если уровень аминокислоты высокий, то ребенку назначается специальное лечение. Такие анализы берутся у всех детей.
Если ребенок страдает фенилкетонурией, и обследование не было вовремя проведено, то проявляются характерные признаки, такие как вялость, судороги, бледность кожи и слишком маленький размер головы. В такой ситуации наличие этих признаков и результатов проведенного исследования крови становятся подтверждением наличия заболевания.
Обратите внимание
Также определить фенилкетонурию можно с помощью генетического анализа, который наиболее часто проводится в случае наличия в семье больного фенилкетонурией. Может потребоваться консультация эндокринолога или медицинского генетика.
Диета, предполагающая ограничение потребления фенилаланина, которой должны придерживаться все больные фенилкетонурией. Такой диеты необходимо придерживаться либо до начала полового созревания человека, либо на протяжении всей жизни. Лечение фенилкетонурией включает в себя:
- ограничение белков животного и растительного происхождения и их замена специальными питательными белковыми смесями;
- для питания детей, страдающих фенилкетонурией, применяются специальные питательные смеси, которые содержат много белка и мало фенилаланина;
- отказ от газированных сладких напитков, жевательных резинок и конфет в состав которых входит фенилаланин;
- лечебная физкультура;
- препараты против судорог, если таковые присутствуют.
Осложнения и последствия
Если у больного фенилкетонурия, лечение которой было начато своевременно, то прогноз достаточно благоприятный. При правильном соблюдении диеты малыш будет нормально расти и развиваться, а никаких повреждений не будет.
В этом случае можно предотвратить опасные последствия в виде нарушений развития головного мозга, разрушений отдельных его клеток – нейронов, а также прочих повреждений.
В редких случаях диетотерапия не является эффективной и не помогает избежать неврологических нарушений – судорог, развития умственной неполноценности.
Профилактика фенилкетонурии
Развитие фенилкетонурии не поддается профилактике, поскольку нельзя предотвратить наличие в организме ребенка дефектного гена.
Если болезнь развивается, то имеет значение только профилактика появления и развития серьезных повреждений мозга путем правильного и своевременного назначения диетотерапии.
С целью пренатальной диагностики (до родов) может быть показано проведение генетического анализа в медико-генетической консультации, что помогает предсказать вероятное развитие заболевания у ребенка. Такие действия проводятся в семьях, где уже были случаи рождения детей с фенилкетонурией.
Фенилкетонурия: признаки, диагностика, лечение

Фенилкетонурия — наследственное заболевание, довольно распространенное. Для этого заболевания характерны нарушения белкового обмена. Самое тяжелое последствие фенилкетонурии — повреждение головного мозга, а также сопровождающие нарушения физического и психического развития ребенка.
Классическая фенилкетонурия, признаки, диагностика, лечение которой описаны ниже, в 1934 году впервые была описана А. Феллингом. А уже в 50 годах в Англии лечение фенилкетонурии прошло успешно.
К концу 60 годов разработали способ обнаружения фенилкетонурии у новорожденных, этот метод получил название «метод Гатри», который заключался в определении в крови новорожденных содержания фенилаланина. Фенилкетонурия встречается как у мальчиков, так и девочек, при этом частота практически одинаковая.
Наследуется фенилкетонурия по аутосомно-рецессивному виду. То есть, у здоровых родителей, носителей измененного гена, ребенок может родиться больной фенилкетонурией. В настоящее время при фенилкетонурии применяют лишь диетологическое лечение.
При своевременном обнаружении заболевания и при правильной диете больной фенилкетонурией ребенок может вырасти абсолютно здоровым.
Типы фенилкетонурии
Существует три типа фенилкетонурии, все они отличаются как по проявлениям, так и по методам лечения.
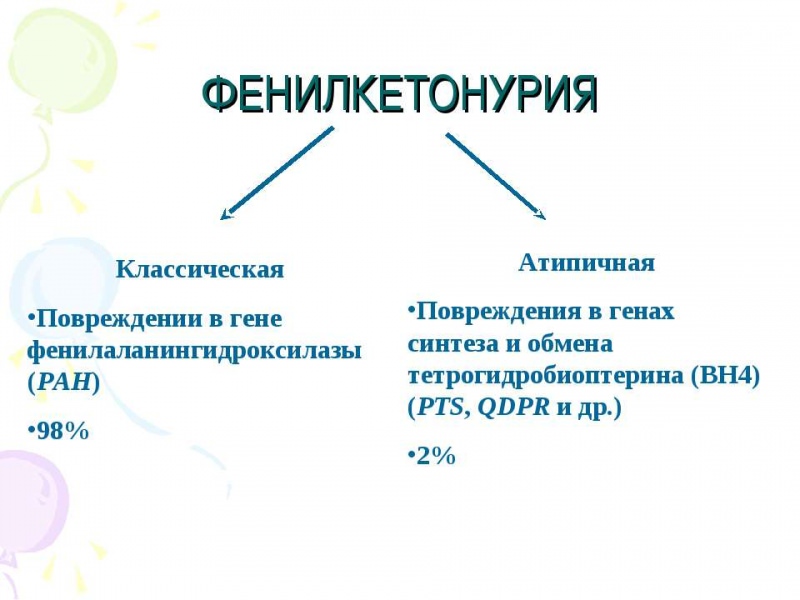
Фенилкетонурия 1-го типа приходится на 98% случаев, так как является самой распространенной классической формой заболевания.
В основе развития этой формы находится недостаток фермента фенилаланин-4-гидроксилаза, который нужен для того, чтобы аминокислоты фенилаланина превращались в тирозин.
Фенилкетонурия 2-го типа приходится на 1-2% случаев. В основе этого типа заболевания лежит недостаток фермента дигидроптеридинредуктазы.
Важно
При этом типе фенилкетонурии доминируют судороги, тяжелая умственная отсталость. Этот тип достаточно быстро прогрессирует, поэтому к 2-3 годам ребенок умирает.
Фенилкетонурия 3-го типа вызывается недостатком тетрагидробиоптерина.
Течение этого типа проходит аналогично второму типу, но с прибавлением микроцефалии — уменьшение объема мозга.
Причины развития фенилкетонурии
- Мутация (изменение) гена, распределенного на двенадцатой хромосоме;
- Близкородственные браки (увеличивают риск рождения больного фенилкетонурией ребенка).
Что наблюдается при фенилкетонурии?
Выше писали, что наблюдается недостаток фермента фенилаланин-4-гидроксилаза, который нужен для того, чтобы аминокислоты фенилаланина превращались в тирозин. Производные фенилаланина, накапливаясь в крови, отравляюще сказываются на нервной системе.
Фенилкетонурия: признаки и симптомы
После рождения в большинстве случаев фенилкетонурия протекает без каких-либо симптомов, поэтому ребенок выглядит абсолютно нормальным. Больные фенилкетонурией дети рождаются в срок, у них нормальный рост и масса тела, зачастую они рождаются со светлой кожей, голубыми глазами, светлыми волосами.
Первые симптомы у новорожденных иногда появляются спустя несколько недель. Основным проявлением этого заболевания у новорожденных является неукротимая рвота, нередко принимаемая за признак сужения выхода из желудка (пилоростеноза).
Основные симптомы проявляются в возрасте двух-шести месяцев: педиатр совместно с родителями отмечает отставание ребенка как в физическом, так и психическом развитии. Такие дети сидеть и ходить начинают с запозданием.
Помимо этого, наблюдается специфический запах мочи и пота, при этом развивается повышенная потливость.
Другие симптомы этого заболевания:
- Упорная рвота;
- Вялость;
- Уменьшение размеров головы;
- Апатия к окружающему миру;
- Беспричинная плаксивость;
- Повышенная раздражительность;
- Беспокойство;
- Позднее прорезывание зубов (начинается после 18 месяцев)
- Судорожные припадки;
- Кожные изменения (дерматиты, экзема);
- Сонливость.
По мере прогрессирования заболевания у ребенка замечается увеличение мышечного тонуса определенных групп мышц, что сопровождается специфической позой больного ребенка, например, позой «портного» — ноги в коленях поджатые, а руки в локтях согнутые. Также замечается дрожание рук.
Начиная с 6 месячного возраста ребенка можно заметить, что он отстает в психическом развитии. Развивается задержка психоречевого развития (это когда ребенок не произносит слоги). Формируется задержка моторного развития (вследствие чего ребенок позже начинает самостоятельно держать головку, сидеть, вставать на ножки, ходить). Если лечение не начать вовремя, то у ребенка развивается глубокая психическая инвалидность. Таким образом, у детей (60%) старше трех-четырех лет замечается практически полное отсутствие мышления и речи.
Фенилкетонурия: диагностика
На 4-5 день жизни доношенного ребенка и на 7 день недоношенного ребенка в роддоме берется кровь на наличие ФКУ. Поставить диагноз фенилкетонурии очень важно сразу при рождении ребенка, это необходимо для профилактики осложнений ФКУ.
Совет
Спустя один час после кормления одной каплей капиллярной крови насыщают специальный бумажный лист. Если концентрация фенилаланина превышает 2,2мг%, то ребенка и родителей для осмотра отправляют в медико-генетический центр, чтобы там полностью обследоваться и уточнить диагноз.
Фенилкетонурия: лечение
Существует единственный способ лечения этого заболевания — диетотерапия, притом начинать нужно сразу после обнаружения заболевания. Диетотерапия заключается в резком ограничении употребления продуктов, содержащих фенилаланин, поэтому исключаются продукты с большим содержанием белка — рыба, мясо, яйцо, творог, крупы. А также бобовые, орехи, шоколад, хлебобулочные изделия.
Как правило, лечебный рацион больного ФКУ состоит из специализированных продуктов как отечественного, так и зарубежного производства. До года лечебные продукты ребенка по составу максимально приближены к грудному молоку, например, такие смеси как «Лофенилак», «Афенилак ». После года применяются другие смеси, например, «Тетрафен», «Максамум-ХР», «Фенил-фри». «Максамум-ХР» предназначенные для детей 6-8 лет и беременным женщинам, больным ФКУ.
Больные ФКУ, как и все, нуждаются в источнике жира, для них этими источниками являются сливочное, топленое, растительное масло. Также в рацион следует включить разные овощи, соки, фрукты, сахара. Такие дети должны постоянно находиться под наблюдением не только педиатра, но и психоневролога.
В начале лечения необходимо еженедельно следить за содержанием фенилаланина, потом, при нормальных показателях, после года анализ берется один раз в два-три месяца.
От срока начала лечения фенилкетонурии будет зависеть прогноз относительно умственного развития.
Фенилкетонурия — самое яркое нарушение обмена аминокислот

При любых нарушениях превращения фенилаланина в тирозин развивается фенилкетонурия.
По Mc Kusick выделяется несколько типов фенилкетонурии: классическая (1 типа), вариантная (2 типа), 3 типа и материнская.
Превращение фенилаланина при фенилкетонурии
Фенилкетонурия 1 типа (классическая)
Фенилкетонурия 1 типа является наиболее распространенной аминоацидопатией. Частота ФКУ среди новорожденных по данным массового скрининга в различных странах составляет в среднем 1:10000, однако значительно варьирует в зависимости от популяции: от 1:4560 в Ирландии, до 1:100000 в Японии.
Этиология
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией, которая вызывает снижение активности фермента фенилаланин-4-монооксигеназы, обеспечивающей превращение фенилаланина в тирозин. Фермент имеется только в печени, почках, поджелудочной железе.
Патогенез
В патогенезе ФКУ имеют значение многие обстоятельства, в частности:
- значительное накопление в тканях и жидкостях больного организма фенилаланина и его производных (фенилпировиноградная, фенилмолочная (миндальная), фенилуксусная, гиппуровая кислоты, фенилэтиламин, фенилацетилглютамин) и вызванный ими ацидоз,
- прямое токсическое действие указанных веществ на центральную нервную систему, которое заключается в торможении фенилаланином активности ряда ферментов, в том числе пируваткиназы (окисление глюкозы), тирозиназы (синтез меланина), тирозин-гидроксилазы (синтез катехоламинов) и нарушение синтеза моноаминовых нейромедиаторов – тирамина, октопамина,
- нарушение синтеза серотонина, т.к. фенилаланин-4-монооксигеназа также вовлечена в гидроксилирование триптофана до 5-гидрокситриптофана, предшественника серотонина,
- конкурентное снижение фенилаланином транспорта в клетки ароматических аминокислот – триптофана и тирозина,
- нарушение синтеза простых и сложных белков в тканях, что вызывает тяжелые повреждения мозга и нарушение функции печени у большинства больных.
Клиническая картина
Ребенок с фенилкетонурией выглядит при рождении здоровым. Манифестация ФКУ происходит на первом году жизни, обычно в возрасте 2-6 мес. Первым симптомом заболевания может стать рвота.
Другими ранними проявлениями болезни служат вялость ребенка, чрезмерная сонливость, отсутствие интереса к окружающему, иногда повышенная раздражительность, беспокойство, плаксивость, также отмечаются срыгивания, нарушение мышечного тонуса (чаще мышечная гипотония), судороги.
Характерным признаком является повышенная потливость, от мочи и пота исходит необычный запах фенилуксусной кислоты, который характеризуют как заплесневелый, мышиный или волчий.
Дети отстают в физическом и нервно-психическом развитии.
Обратите внимание
Задержка психического и речевого развития может происходить постепенно и стать очевидной лишь через несколько месяцев. Нелеченный ребенок теряет около 50 баллов IQ к концу первого года жизни. Только 4-5% остаются на стадии дебильности, остальные имеют коэффициент интеллекта
Фенилкетонурия

Фенилкетонурия – тяжелое наследственное заболевание, которое характеризуется главным образом поражением нервной системы. Заболевание наследуется по аутосомно-рецессивному типу.
Признаки фенилкетонурии обнаруживаются уже в первые недели и месяцы жизни. Дети
отстают в физическом и нервно-психическом развитии; отмечаются вялость, чрезмерная сонливость или повышенная раздражительность, плаксивость.
По мере прогрессирования болезни могут наблюдаться эпилептиформные припадки – развернутые судорожные и бессудорожные типа кивков, поклонов, вздрагиваний, кратковременных отключений сознания. Гипертония отдельных групп мышц проявляется своеобразной «позой портного» (поджатые ноги и согнутые руки).
Могут наблюдаться гиперкинезы, тремор рук, атаксия, иногда парезы по центральному типу. Дети нередко белокурые со светлой кожей и голубыми глазами, у них часто отмечаются дерматиты, экзема, повышенная потливость со специфическим (мышиным) запахом пота и мочи. Обнаруживается склонность к артериальной гипотензии.
При отсутствии лечения развивается идиотия или имбецильность, глубокая психическая инвалидность.
Диагностика
Чрезвычайно важно установить диагноз в доклинической стадии или, по крайней мере, не позднее 2-го месяца жизни, когда могут проявиться первые признаки болезни.
Для этого всех новорожденных обследуют по специальным программам скрининга, выявляющего повышение концентрации фенилаланина в крови уже в первые недели жизни.
Каждого ребенка, у которого обнаруживаются признаки задержки развития или минимальная неврологическая симптоматика, необходимо обследовать на патологию обмена фенилаланина.
Важно
Используют микробиологический и флюорометрический методы определения концентрации фенилаланина в крови, а также пробу Фелинга на фенилпировиноградную кислоту в моче (прибавление нескольких капель 5% раствора треххлористого железа и уксусной кислоты к моче больного приводит к появлению зеленой окраски пятна на пеленке). Эти и другие подобные методы относятся к категории ориентировочных, поэтому при положительных результатах требуется специальное обследование с использованием точных количественных методов определения содержания фенилаланина в крови и моче (хроматография аминокислот, использование аминоанализаторов и др.), которое осуществляется централизованными биохимическими лабораториями.
Дети требуют специального наблюдения и лечения в медико-генетических центрах (кабинетах поликлиник).
Дифференциальный диагноз проводят с внутричерепной родовой травмой, внутриутробными инфекциями.
Лечение
При подтверждении диагноза биохимическими методами необходим перевод детей на специальную диету, с ограничением фенилаланина, что при ранней диагностике гарантирует нормальное нервно-психическое развитие ребенка.
Молоко и другие продукты животного происхождения из диеты исключают и назначают белковые гидролизаты, которые становятся главными продуктами питания, обеспечивающими потребность в белке. Белковые гидролизаты вводят с фруктовыми и овощными соками, пюре, супами.
Лечение проводят под контролем содержания фенилаланина в крови, добиваясь поддержания его уровня в пределах 0,03-0,04 г/л. Строгое ограничение белков животного происхождения требуется на протяжении первых 2-3 лет жизни
Профилактика
Большое значение имеет специальное наблюдение за семьями риска, т. е. за такими семьями, где уже имелись дети с фенилкетонурией.
Новорожденные из этих семей должны быть подвергнуты обязательному биохимическому исследованию и при показаниях – раннему лечению.
Выявление и лечение детей по программам массового скрининга также позволяет предупредить развитие тяжелой психической инвалидности.
Предложен ДНК-зонд для пренатальной диагностики фенилкетонурии в семьях высокого риска.
© Большая медицинская энциклопедия
Фенилкетонурия

Фенилкетонурия известна с 1934 года, но вследствие отсутствия качественной диагностики заболевание выявлялось только на той стадии, когда в результате токсичного воздействия на головной мозг нарушения умственного развития становятся необратимыми. Именно поэтому заболевание получило второе название — фенилпировиноградная олигофрения.
По статистике нарушение обмена фенилаланина чаще наблюдается у девочек.
Распространено больше в Турции, северных странах Европы (исключение – Финляндия), и крайне редко встречается у представителей негроидной расы.
Поскольку фенилкетонурия относится к заболеваниям, при которых дефективный ген наследуется от обоих родителей, браки между родственниками увеличивают вероятность рождения детей, страдающих этим заболеванием.
Совет
Впервые имевшее успех лечение провели в детском госпитале Бирмингема (Англия) в 50-х гг. XX века, но по-настоящему эффективным этот метод лечения стал после внедрения в начале 60-х гг. ранней диагностики.
Фенилаланин под воздействием ферментов в организме преобразуется в тирозин – аминокислоту, которая выводится из организма. В зависимости от дефекта гена, блокирующего определенный фермент, выделяют:
- Фенилкетонурию I-го типа (классическую или тяжелую). Вызывается генной мутацией, нарушающей выработку печеночного фермента фенилаланингидроксилазы и преобразование фенилаланина в тирозин.
- Фенилкетонурию II-го типа (атипичную). Характеризуется генным дефектом, вызывающим недостаток дигидробиоптерин-редуктазы. Благодаря этому фактору нарушается восстановление активности органического соединения, необходимого для преобразования фенилаланина. Также наблюдается пониженное содержание в спинномозговой жидкости и в сыворотке крови витамина В9, необходимого для утилизации аминокислот.
- Фенилкетонурию III-го типа (атипичную). Провоцируется нехваткой катализатора, необходимого для синтеза тетрагидробиоптерина (нужен для преобразования фенилаланина в тирозин).
- Примаптеринурию — атипичную форму, которая наблюдается при легкой форме гиперфенилаланинемии. Ферментный дефект этого вида ФКУ в настоящее время не выяснен, но данная форма заболевания характеризуется значительным количеством примаптерина и его производных в моче, а количество нейромедиаторных метаболитов в спинномозговой жидкости не отклоняется от нормы.
Выделяется также материнская фенилкетонурия, наблюдающаяся у потомства женщин, больных ФКУ и не соблюдающих специализированную диету. Патогенез данной формы заболевания до конца не изучен, но отмечается, что без постоянного контроля за уровнем фенилаланина у новорожденных обнаруживается ряд патологических изменений:
- недостаточный вес мозга;
- увеличенные в размерах желудочки головного мозга (вентрикуломегалия);
- гипоплазия (недоразвитие) белого вещества и задержка миелинизации.
Фенилкетонурия данного типа вызывает хроническую интоксикацию плода и приводит к умственной отсталости ребенка.
Фенилпировиноградная олигофрения возникает при наличии патологического гена у матери и отца. Тип наследования — аутосомно-рецессивный, то есть не зависит от пола ребенка. Вероятность развития заболевания у новорожденных, родители которых являются носителями дефектного гена, составляет 25%.
В результате вызванного генным дефектом нарушения обмена фенилаланина и его последующего превращения в тирозин в организме накапливаются токсические производные этой аминокислоты (фенилпировиноградная, фенилмолочная и фенилуксусная кислоты), в норме встречающиеся в минимальном количестве. Образуются также не встречающиеся в норме ортофенилацетат и фенилэтиламин, нарушающие липидный обмен в головном мозге, что служит причиной прогрессирующего снижения интеллекта.
Факторами, влияющими на развитие патологии, являются:
- нарушения аминокислотного обмена;
- нарушения миелинизации;
- нарушение синтеза протеолипидных белков,
- пониженный уровень нейротрансмиттеров (адреналина, серотонина и др.).
Сразу после рождения у детей с ФКУ I симптомы болезни не наблюдаются, хотя обычно имеется специфический ряд внешних признаков:
В возрасте 2-х – 6-ти месяцев проявляются первые признаки заболевания:
- вялость (не делает попыток перевернуться, сесть);
- пассивное восприятие окружающего (не реагирует на мать, не отвечает улыбкой);
- повышенная раздражительность;
- задержка психомоторного развития.
Возможна интенсивная частая рвота и беспокойство, приступы судорог.
Если заболевание своевременно не выявлено и в рацион ребенка вводят белковую пищу, симптоматика начинает нарастать. У таких детей поздно прорезываются зубы, а череп несколько уменьшен в размерах. Сидеть и ходить начинают позже сверстников, мимика невыразительна, в год не способны выразить голосом эмоции, речь взрослых не понимают. Возможна задержка роста.
Поскольку фенилаланин в организме не преобразуется, он выводится с потом и мочой, поэтому от больных исходит затхлый или «мышиный» запах.
Фенилкетонурия у детей проявляется также в своеобразных позах и походке, так как мышечный тонус у таких больных повышен. В положении стоя ноги у ребенка широко расставлены и согнуты в коленях и тазобедренных суставах, голова и плечи опущены. Походка качающаяся, шаги мелкие. Сидят они в положении портного (поджимают и скрещивают ноги).
Фенилпировиноградная олигофрения после трехлетнего возраста проявляется в:
- повышенной возбудимости и утомляемости;
- нарушениях поведения;
- психотических расстройствах;
- умственной отсталости.
Часто наблюдается экзема, склеродермия и дерматит.
При отсутствии лечения состояние больных ухудшается. Своевременно поставленный диагноз и лечение позволяют предотвратить нарушения развития ребенка.
Диагностировать заболевание можно:
- полуколичественным тестом, который позволяет определить приблизительное количество фенилаланина в крови;
- количественным определением при помощи реагентов, выявляющих количество фенилаланина в высохших кровяных пятнах.
Для этого обследования в роддоме в день выписки или на 5-й день жизни у всех детей после кормления берется кровь из пятки.
Забор крови у двухнедельного ребенка для теста на фенилкетонурию
Диагностика включает и исследование мочи (проба Феллинга), но оно информативно только после 10-12 дня жизни ребенка. Фенилпировиноградная кислота проявляется сине-зеленой окраской в моче при добавлении в мочу хлорида железа.
Можно также определить активность фермента фенилаланингидроксилазы в материале, полученном методом биопсии, и обнаружить мутации в гене.
Используются также такие методы, как:
- тест Гатри, основанный на добавлении крови в бактериальную культуру, быстро растущую в фенилаланиновой среде;
- тонкослойная хроматография аминокислот, содержащихся в сыворотке крови;
- флуориметрия, позволяющая выявить микродозы фенилаланина благодаря облучению ультрафиолетом.
Фенилкетонурия 2 и 3 типа диагностируется при помощи исследования биоптеринов мочи, перорального нагрузочного теста с тетрагидробиоптерином, энзиматических исследований.
Выявлять фенилкетонурию можно и с помощью генетического анализа, который обычно проводят при наличии членов семьи с данным диагнозом.
При своевременно поставленном диагнозе лечение фенилкетонурии заключается в специализированной диете, ограничивающей поступление пищи, содержащей фенилаланин. Так как все естественные источники белка содержат около 4% фенилаланина, они заменяются содержащими другие аминокислоты синтетическими продуктами. Наиболее эффективна диета, назначенная до 8-й недели жизни.
Грудным детям назначаются полностью очищенные от лактозы смеси на основе гидролизата молочного белка. Материнское молоко допустимо в ограниченном количестве.
Хотя раньше врачи рекомендовали соблюдать диету только до окончания развития головного мозга (20 лет), повышение уровня фенилаланина после прекращения диеты у многих людей вызывает психиатрические проблемы. Фенилкетонурия у взрослых проявляется недостаточностью мотивации, бессонницей и деконцентрацией, импульсивностью и т.д., поэтому диету рекомендуется соблюдать пожизненно.
Диетические ограничения, введенные после 2-х лет, способны только снизить выраженность симптомов.
Для лечения применяются также препараты:
- относящиеся к ноотропной группе (ноотропил и др.). Они стимулируют умственную деятельность и активизируют когнитивные функции;
- содержащие витамины, аминокислоты и белки («Афенилак» и т.д.).
Атипичные формы фенилкетонурии не поддаются диетотерапии и требуют дополнительного медикаментозного лечения, которое включает:
- Дигидробиоптерин, компенсирующий недостаток фолиевой кислоты;
- Леводол, действующий на ЦНС.
Пациентам, нуждающимся в дополнительной коррекции нейромедиаторов, назначают «Мадопар» или «Наком».
Взрослым назначаются растительные препараты, содержащие фенилаланингидроксилазу.
Поскольку фенилкетонурия – генетическое заболевание, полностью предотвратить его развитие нельзя. Профилактические меры направлены на предотвращение необратимых тяжелых нарушений развития мозга путем своевременной диагностики и диетотерапии.
Семьям, в которых уже были случаи данного заболевания, рекомендуется проведение генетического анализа, способного предсказать возможное развитие фенилкетонурии у ребенка.
Обратите внимание
Женщинам с ФКУ рекомендуется до зачатия и во время беременности придерживаться диеты с низким содержанием фенилаланина.
Фенилкетонурия

Фенилкетонурия – наиболее распространенное нарушение обмена аминокислот. В среднем фенилкетонурии подвержен 1 из 8000 человек.
В основе болезни лежит дефицит фермента, осуществляющего превращение фенилаланина в тирозин (тирозин препятствует отложению жиров, снижает уровень аппетита, улучшает функции гипофиза, щитовидной железы и надпочечников).
Симптомы фенилкетонурии
Фенилкетонурия проявляется на первом году жизни. Основными симптомами в этом возрасте являются:
- вялость ребенка;
- отсутствие интереса к окружающему;
- иногда повышенная раздражительность;
- беспокойство;
- срыгивания;
- нарушения мышечного тонуса (чаще мышечная гипотония);
- судороги;
- признаки аллергического дерматита;
- появляется характерный «мышиный» запах мочи.
В более позднем возрасте для больных фенилкетонурией характерна задержка психоречевого развития, нередко отмечается микроцефалия.
При фенилкетонурии характерны следующие фенотипические особенности: гипопигментация кожи, волос, радужной оболочки глаз. У некоторых больных одним из проявлений патологии может быть склеродермия.
Эпилептические приступы встречаются почти у половины больных фенилкетонурией и в некоторых случаях могут служить первым признаком болезни.
Диагностика
Диагноз при подозрении на фенилкетонурию основывается на совокупности генеалогических данных, результатов клинического и биохимического обследования:
- возможный родственный брак родителей больного ребенка;
- аналогичная патология у родных или двоюродных сибсов (братьев или сестер);
- судороги, нарушение мышечного тонуса;
- экзематозные изменения кожи;
- гипопигментация волос, кожи, радужной оболочки глаз;
- своеобразный «мышиный» запах мочи;
- повышенный уровень фенилаланина в крови > 900 мкмоль/л;
- присутствие в моче фенилпировиноградной, фенилмолочной, фенилуксусной кислот;
- положительная проба Феллинга.
В настоящее время для диагностики фенилкетонурии разработаны и внедрены молекулярно-генетические методы выявления генного дефекта.
Диагностика у новорожденных (скрининг)
В связи с достаточной распространенностью фенилкетонурии, тяжестью клинических проявлений и реальной возможностью профилактического лечения, фенилкетонурия в числе первых наследственных нарушений обмена веществ была включена в список наследственных заболеваний, рекомендованных Всемирной организацией здравоохранения для раннего выявления среди новорожденных.
Для ранней диагностики фенилкетонурии в России осуществляется массовое обследование детей в родильных домах с определением уровня фенилаланина в крови.
Забор крови производится у новорожденных в возрасте 4–5 дней. Реже объектом исследования является моча.
Лечение фенилкетонурии
Главным способом лечения фенилкетонурии является диетотерапия, ограничивающая поступление в организм белка и фенилаланина.
Основным критерием адекватности диеты при фенилкетонурии служит уровень фенилаланина в крови, который должен:
- в раннем возрасте составлять 120–240 мкмоль/л;
- у детей дошкольного возраста – не превышать 360 мкмоль/л;
- у школьников – не превышать 480 мкмоль/л;
- у детей старшего школьного возраста допустимо увеличение содержания фенилаланина в крови до 600 мкмоль/л.
Пищевой рацион строится путем резкого ограничения поступления белковых продуктов животного и растительного происхождения и, следовательно, фенилаланина. Для облегчения расчетов принято считать, что 1 г условного белка содержит 50 мг фенилаланина.
При лечении фенилкетонурии полностью исключают продукты, богатые белком и фенилаланином: мясо, рыбу, сыр, творог, яйца, бобовые и др. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты – амилофены.
Для коррекции белкового питания и восполнения недостатка аминокислот при фенилкетонурии назначаются специальные лечебные продукты:
- белковые гидролизаты: нофелан (Польша), апонти (США), лофенолак (США);
- смеси L-аминокислот, лишенные фенилаланина, но содержащие все другие незаменимые аминокислоты: фенил-фри (США), тетрафен (Россия), П-АМ универсальный (Великобритания).
Несмотря на обогащение аминокислотных смесей и белковых гидролизатов минеральными и другими веществами, больные фенилкетонурией нуждаются в дополнительном назначении витаминов, в частности группы В, минеральных соединений, особенно содержащих кальций и фосфор, препаратов железа и микроэлементов.
В последние годы для страдающих фенилкетонурией была обоснована необходимость применения препаратов карнитина (L-карнитин, элькар в средней суточной дозе 10–20 мг/кг массы в течение 1–2 мес. 3–4 курса в год) для профилактики его недостаточности.
Параллельно лечение фенилкетонурии осуществляется медикаментозным патогенетическим и симптоматическим лечением ноотропными средствами, препаратами, улучшающими сосудистую микроциркуляцию, по показаниям – антиконвульсантами.
Важно
Широко используется лечебная гимнастика, общий массаж и др. Комплексная реабилитация детей с фенилкетонурией предусматривает специальные методы педагогических воздействий в процессе подготовки к школе и школьного обучения. Больные нуждаются в помощи логопеда, педагога, в ряде случаев – дефектолога.
Большие споры вызывает вопрос о длительности диетотерапии в лечении фенилкетонурии.
В последнее время большинство врачей принимает точку зрения о необходимости продолжительного выполнения диетических рекомендаций.
Обследование детей, прекративших соблюдать диету в школьном возрасте, и детей, продолжавших получать диетотерапию, однозначно показало значительно более высокий уровень интеллектуального развития последних.
У больных фенилкетонурией старшего возраста, в том числе подростков, безусловно, возможно постепенное расширение диеты в связи с улучшением толерантности к фенилаланину.
Коррекция питания осуществляется, как правило, путем введения в рацион ограниченного количества круп, молока и некоторых других натуральных продуктов, содержащих относительно умеренное количество фенилаланина.
В период расширения рациона проводятся оценка нервно-психического статуса детей, контроль электроэнцефалограммы, уровня фенилаланина в крови.
В возрасте старше 18–20 лет проводится дальнейшее расширение диеты, однако и во взрослом периоде пациентам рекомендуется отказаться от высокобелковых продуктов животного происхождения.
Совет
Особенно строго подходят к диетотерапии девочек, страдающих фенилкетонурией, и женщин в репродуктивном периоде. Такого рода больным фенилкетонурией необходимо продолжать диетическое лечение для обеспечения рождения здорового потомства.
В последние годы разрабатывается способ снижения уровня фенилаланина в крови путем приема препарата, содержащего фенилаланингидроксилазу растительного происхождения.
Данная статья опирается на статью из книги «Врожденные и наследственные заболевания» под редакцией профессора П.В.Новикова, М., 2007
Comments
(0 Comments)