Атаксия мозжечковая, вестибулярная, телеангиэктазия и статическая, поздняя наследственная пьера мари









Проверяется также фланговая походкашаговые движения в сторону. При этом обращают внимание на четкость шага и возможность быстрой остановки при внезапной команде. В случае поражения мозжечковых систем при этих исследованиях выявляется нарушение походки описанного характера. Такая походка называется атактической или мозжечковой. Также нарушается сочетание простых движений, составляющих последовательную цепь сложных двигательных актовасинергия или диссинергия.
Что касается топической диагностики поражения мозжечка, то следует сказать, что если нарушается координация движений при стоянии и ходьбе, то это поражение ростральной части червя мозжечка. А если туловищная атаксия нарушения равновесия при сидении, то возникает поражение каудальной части червя мозжечка.
Асинергия определяется, в частности, с помощью пробы Бабинского.
Также исследуется: – симптом Ожеховского; -симптом отсутствия «обратного толчка Стюарта-Холмса» ; При динамической атаксии исследуются: -ПНП; -ПКП; -проба на диадохокинез; -проба на соразмерность движений; -указательная проба; -проба Шильдера;
Нарушение мелкой моторики при письме, также характерно для поражения коры мозжечка
Диагностика заболевания основывается на клинической картине и данных МРТ исследования. Лечение симптоматическое. Прогноз заболевания относительно благоприятный.
БЛАГОДАРЮ ЗА ВНИМАНИЕ
Атаксия мозжечковая, вестибулярная, телеангиэктазия и статическая, поздняя наследственная Пьера Мари
Атаксия является серьезным заболеванием, провоцирующим расстройство моторики. Патология сопровождается нарушением координации движений. Атаксия развивается вследствие поражения определенных участков мозга.
Виды атаксии
Атаксия классифицируется в зависимости от того, какая часть мозга поражена. Так, различают следующие виды нарушений:
- сенситивное нарушение;
- мозжечковое;
- вестибулярное;
- корковое.
При сенситивной форме, нарушение происходит на уровне проводников глубокомышечной чувствительности. Поражение мозжечка называется мозжечковой атаксией. Вестибулярная атаксия развивается вследствие нарушений вестибулярного аппарата пациента. Корковая или лобная атаксия возникает вследствие поражения соответствующей части мозга.
Симптомы атаксии зависят от того, какая часть мозга повреждена.
Симптомы сенситивной атаксии
Эта форма заболевания развивается из-за поражения рецепторов, отвечающих за чувствительность мышц. Причиной нарушения становятся патологические процессы центральной нервной системы.
Причинами патологии могут выступать:
- полинейропатия;
- поражения задних нервов;
- поражения спинного мозга;
- поражение коры головного мозга.
Заболевание развивается на фоне оперативного вмешательства в головной мозг при необходимости удаления опухолей. Также поражение спинного мозга может быть спровоцировано вирусными и инфекционными заболеваниями в запущенной стадии. Наиболее распространенная причина сенситивной атаксии – это инсульт.
Симптомами сенситивного синдрома является:
- ослабление мышечного тонуса;
- потеря веса;
- нарушение сгибательной функции всех суставов;
- нарушение моторики;
- рефлекторные нарушения;
- изменения походки.
Прогрессирование заболевания приводит к невозможности самостоятельного передвижения и инвалидности.
Лечение сенситивной формы болезни
Лечение во многом зависит от причины нарушения. Основу лечения составляет терапия патологии, которая привела к нарушению моторики. Так, при вирусных и инфекционных заболеваниях обязательно назначаются антибиотики и противовирусные препараты.
Лечение основано на использовании следующих препаратов:
- витамины группы В;
- инъекции рибофлавина и иммуноглобулина;
- ноотропные препараты;
- антихолинэстеразные препараты.
Ноотропные препараты эффективны при сосудистых заболеваниях, которые стали причиной развития атаксии. Лекарства этой группы применяются для улучшения мозгового кровообращения и укрепления нервной системы. Препараты способствуют стимуляции мозговой активности.
Антихолинэстеразные препараты показаны для лечения в том случае, если патология сопровождается невритами и дистрофией мышц. Лекарства этой группы помогают улучшить проведение импульсов в нервные окончания. Наряду с медикаментозной терапией показано лечение физкультурой и физиотерапевтические методы. Гимнастика способствует улучшению моторики, но только при условии регулярного выполнения.
Мозжечковая и корковая форма нарушения
Эта форма нарушения развивается из-за повреждения мозжечка. Патологию вызывают следующие причины:
- интоксикация организма тяжелыми металлами и токсическими веществами;
- ишемия мозга;
- энцефалит;
- опухоли;
- вирусные заболевания.
Кроме того, существует генетически обусловленная форма мозжечковой атаксии, которая встречается у детей.
Поражение мозжечка и нарушение моторики бывает двух видов – это динамическая атаксия и статико-локомоторное нарушение. Статическая атаксия (локомоторная атаксия) характеризуется нарушением равновесия, когда пациент стоит, в то время как при динамической форме происходит нарушение равновесия во время ходьбы.
При мозжечковой атаксии симптомы следующие:
- проблемы с равновесием;
- падения;
- нарушения походки;
- нарушения речи;
- снижение тонуса мышц.
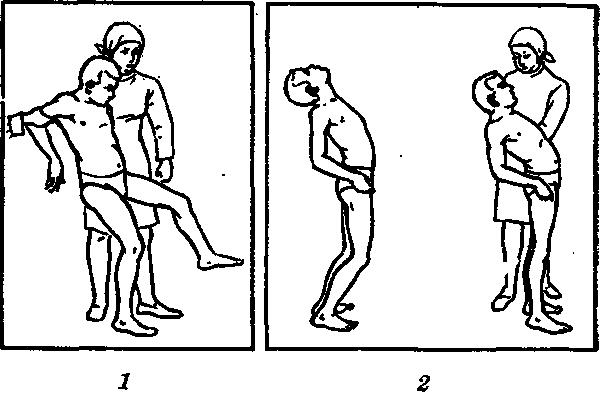
Проблемы с равновесием выражаются выраженным перекосом на пораженную сторону, при этом возможны внезапные падения при ходьбе. Нарушения походки выражаются стремлением ходить, широко расставив ноги. Походка шаркающая, раскачивающаяся, туловище напряжено. Ходьба по прямой сильно затруднена.
Возможны нарушения речи, которые проявляются медлительностью и неуверенностью. Часто пациенты делают ударения на каждом слоге.
При поражении коры головного мозга, развивается корковая атаксия. Характерными симптомами этой патологии является изменение походки пациента. Походка больных неустойчивая, с тенденцией отклонения назад.
Возможны нарушения, вызывающие временный паралич, когда пациент не может самостоятельно ходить либо стоять.
Заболевание также может сопровождаться галлюцинациями, изменением мышления и другими симптомами поражения коры головного мозга.
Лечение мозжечковой атаксии
При мозжечковой атаксии лечение носит симптоматический характер и подбирается индивидуально для каждого пациента.
Начало терапии начинается с симптоматического лечения заболевания, послужившего толчком к развитию атаксии. При необходимости, проводится детоксикация организма. Далее применяются препараты для улучшения мозгового кровообращения, седативные средства для снятия гипертонуса мышц и укрепления нервной системы, ноотропные препараты.
Лечение включает прием витаминов группы В и минеральных комплексов, которые укрепляют нервную систему и улучшают прохождение импульсов по нервным волокнам. Для улучшения моторной функции показан курс ЛФК.
Если атаксия спровоцирована сосудистыми заболеваниями или действием инфекции, прогноз, как правило, благоприятный. Наиболее неблагоприятный прогноз относится к мозжечковой атаксии, которая развивается на фоне злокачественных опухолей мозга.
Причины и симптомы вестибулярной атаксии

Вестибулярная атаксия развивается вследствие поражения вестибулярного аппарата, что может быть обусловлено инфекционными заболеваниями среднего уха. Причинами заболевания являются травмы уха, злокачественные опухоли, острые гнойные воспаления, хронический отит.
Вестибулярная атаксия характеризуется следующими симптомами:
- неуверенная походка;
- головокружение при повороте головы;
- вегетативные нарушения;
- панические атаки.
Больные передвигаются медленно и осторожно, постоянно испытывая чувство вращения окружающих предметов. Любое резкое движение может вызвать перекос тела пациента в сторону пораженного уха. Походка шаткая, шаркающая. Поворот головы сопровождается сильным головокружением и дезориентацией. На этом фоне развиваются проблемы со сном, возможны внезапные панические атаки.
Вестибулярная атаксия сопровождается вегетативными нарушениями — тахикардией, тошнотой, бледностью кожи лица.
Этот синдром не вызывает нарушения движений конечностей, однако может сопровождаться снижением слуха.
Вестибулярная атаксия лечится симптоматически, терапия основана на лечении первопричины – поражения вестибулярного аппарата или воспалительных процессов среднего уха.
Синдром Пьера-Мари

Синдром моторного нарушения может носить генетически обусловленный характер. В этом случае заболевание вызывают генетические мутации.
Наследственная мозжечковая атаксия Пьера-Мари характеризуется дегенерацией мозжечка, однако первые симптомы заметны в возрасте после 20 лет. Такая мозжечковая атаксия у детей не проявляется. Эта болезнь передается по наследству. Ген, вызывающий дегенеративные процессы в мозжечке, передается ребенку в случае, если хоть один родитель имеет этот синдром.
Для появления первых симптомов патологии необходимо действие какого-либо фактора, в роли которого выступают инфекционные заболевания, беременность, интоксикация организма. Симптомы патологии:
- мозжечковая атаксия;
- нарушения зрения;
- парез лицевого нерва;
- депрессивный невроз;
- снижение интеллектуальных способностей;
- нарушения памяти.
Заболевание постоянно прогрессирует, симптомы усугубляются и это приводит к инвалидности.
Синдром Луи-Бар
Еще одна форма наследственной патологии – это атаксия Луи-Бар. Синдром Луи-Бар проявляется у детей в раннем возрасте и также известен как атаксия телеангиэктазия. Клинические симптомы появляются в возрасте от нескольких месяцев до трех лет, однако становятся заметны, когда ребенок начинает учиться ходить.
Для синдрома характерны следующие признаки:
- проблемы с равновесием;
- тремор во время движения;
- невнятная речь;
- косоглазие;
- отсутствие сухожильных рефлексов;
- снижение мышечного тонуса.
Из-за раннего проявления симптомов дети часто не могут ходить, настолько сильно выражено мозжечковое поражение при синдроме Луи-Бар.
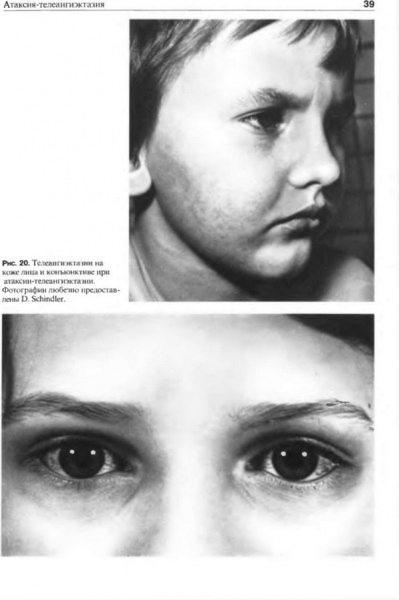
С прогрессирование болезни появляются симптомы телеангиэктазии, которые выражаются образованием сосудистых звездочек по всем телу. В раннем возрасте расширение мелких сосудов локализовано преимущественно на глазных яблоках. Помимо расширенных сосудов, у больных часто наблюдается гиперпигментация кожи, а также белые пятна, веснушки и ранняя седина.
Синдром Луи-Бар вызывает иммунные нарушения, которые приводят к частым инфекционным заболеваниям дыхательных путей. На фоне сниженного иммунитета, любая инфекция может убить человека с этим синдромом.
У больных часто развиваются онкологические заболевания, терапия которых осложнена общим состоянием здоровья пациента.
Патология не лечится, больные редко доживают до 20 лет. Причиной того, что пациенты гибнут в раннем возрасте, являются осложнения синдрома, провоцирующие легочные инфекции и злокачественные новообразования.
Наследственные заболевания, такие как атаксия Пьера-Мари, не имеют специфического лечения. Терапия в этом случае включает прием витаминов, лекарств для стимуляции мозгового кровообращения и мануальную терапию для улучшения мышечного тонуса.
Прогноз
Прогрессирующая патология может стать причиной внезапных падений, что приводит к травмам, из-за чего пациенты могут даже погибать. Поздняя мозжечковая атаксия (туловищная атаксия) приводит к инвалидности. Лечение нужно начинать при появлении первых симптомов.
Прогноз во многом зависит от причины нарушения. Если заболевание было спровоцировано воспалительными процессами и инфекциями, лечение позволяет добиться устойчивой ремиссии.
Успешность излечения во многом зависит от самого пациента, так как наряду с медикаментозной терапией применяются методы ЛФК и массажа.
При регулярном выполнении упражнений моторная функция восстанавливается, однако для этого требуется длительный курс гимнастики.
Наследственные заболевания не лечатся. При болезни Пьера-Мари возможно отсрочить наступление полной недееспособности пациента медикаментозными средствами и гимнастикой, в то время как дети, больные синдром Луи-Бар часто не доживают до совершеннолетия из-за нарушений иммунитета и онкологических заболеваний.
При появлении первых симптомов необходимо пройти тщательное обследование у невролога. Только после анализа результатов диагностики можно судить о степени поражения мозга и дальнейшем прогнозе.
Наследственная мозжечковая атаксия Пьера Мари: причины и симптомы

- 29 Июня, 2018
- Неврология
- Евдокимова Ирина
Мозжечковая атаксия Пьера-Мари — это тяжелое наследственное заболевание. Но в отличие от многих других генетических патологий, недуг впервые проявляется не в детском возрасте, а у взрослых людей. С течением времени симптомы нарастают, а заболевание прогрессирует.
Пути наследования патологии
Наследственная мозжечковая атаксия Пьера-Мари передается детям от больного родителя. При этом вероятность рождения ребенка с патологией составляет 50%. Заболеванию одинаково подвержены и девочки, и мальчики.
Если в семье родился ребенок с таким заболеванием, то в будущем он может передать недуг своим детям. Опасность патологии заключается в том, что на момент деторождения человек может даже не подозревать о том, что он болен.
Первые признаки мозжечковой атаксии Пьера-Мари проявляются в довольно зрелом возрасте. Здоровые дети не являются носителями дефектного гена и не передают заболевание потомству.
Такой вид наследования генетики называют аутосомно-доминантным.

Причины заболевания
Непосредственными причинами мозжечковой атаксии Пьера-Мари являются патологические изменения в коре и ядрах мозжечка. Этот орган в организме человека отвечает за равновесие тела и обеспечивает координацию движений при перемещении.
У больного человека клетки мозжечка подвергаются дегенерации. В результате орган уменьшается в объеме.
Такие же изменения претерпевают и другие отделы центральной нервной системы, например, варолиев мост, который отвечает за передачу спинальных импульсов.
Дегенеративные процессы происходят в продолговатом мозге и в проводящих путях спинного мозга. Заболевание поражает практически все системы организма, отвечающие за равновесие и движение тела.
На поздних стадиях болезни поражаются глазные нервы, что приводит к ухудшению зрения.
Как проявляется болезнь?
Симптомы мозжечковой атаксии Пьера-Мари возникают обычно после 30-ти лет. Можно выделить следующие признаки патологии:
Такая характеристика мозжечковой атаксии Пьера-Мари говорит о том, что это очень серьезное и тяжелое заболевание. Оно неуклонно прогрессирует. Течение патологии усугубляют инфекционные болезни, умственное или физическое напряжение.

Диагностика болезни
При появлении признаков атаксии необходимо обратиться к неврологу. При внешнем осмотре больного выявляется нистагм и другие глазодвигательные отклонения. Заметны речевые нарушения.
Врач проводит пальце-носовую пробу: больному предлагают с закрытыми глазами дотронуться пальцем до кончика носа. Пациент с атаксией обычно не может попасть в цель.
При постукивании неврологическим молоточком по сухожилию определяются повышенные рефлексы.
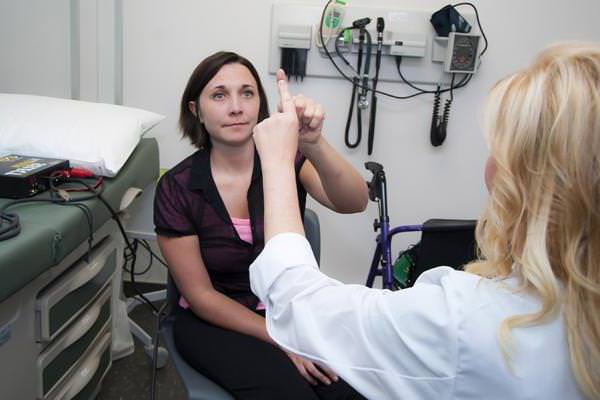
Врач может назначить дополнительные виды обследований, чтобы выявить происхождение атаксии:
- МРТ и КТ головного мозга;
- УЗИ головного мозга.
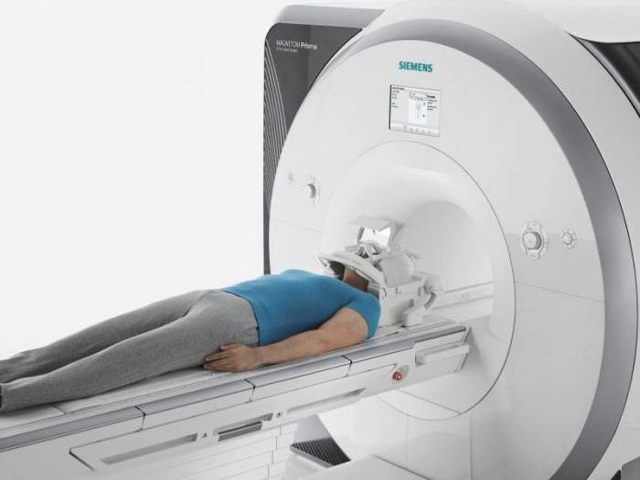
В результатах исследований видна атрофия мозжечка. Кроме этого, врач собирает анамнез. Необходимо уточнить, встречались ли у членов семьи подобные заболевания. Это поможет сделать вывод о наследственном характере атаксии.
Выявить нарушения координации движений несложно. Труднее отделить мозжечковую атаксию Пьера-Мари от других подобных болезней.
Дифференциальный диагноз
Существует множество мозжечковых атаксий и других болезней, сопровождающихся расстройством координации движений, речи и зрения. Врачу необходимо провести дифференциальную диагностику этих состояний.
Похожие проявления встречаются при другом наследственном заболевании – атаксии Фридрейха. Но эта патология проявляется в более молодом возрасте. При болезни Пьера-Мари повышены сухожильные рефлексы, а при атаксии Фридрейха – снижены. Отличается и тип наследования недуга. Болезнь Фридрейха может передаваться потомкам от вполне здоровых родителей, которые являются носителями дефектных генов.
Другим схожим заболеванием является рассеянный склероз. Но эта патология тоже проявляется в более молодом возрасте. Отличить эти две болезни поможет МРТ-обследование. При рассеянном склерозе будут видны очаги демиелинизации нейронов мозга. Этого не наблюдается при атаксии Пьера-Мари.
Излечимо ли заболевание?
На сегодняшний день это заболевание является неизлечимым. Невозможно подобрать такую терапию, которая остановила бы процесс дегенерации клеток мозжечка. Врач может предложить только симптоматическое лечение. Применяются следующие виды препаратов:
Эти медикаменты не способны полностью излечить мозжечковую атаксию Пьера-Мари. Они помогут лишь смягчить симптоматику и замедлить развитие болезни.
Лечебная гимнастика
Больным назначают лечебную гимнастику для укрепления мышц и улучшения скоординированности движений.
Пациентам с атаксией Пьера-Мари полезны упражнения на равновесие. Сначала задания выполняют в сидячем положении. Это может быть тренировка точности движений, повороты тела, подъем и перемещение предметов. Постепенно упражнения усложняют, уменьшая плоскость опоры сидения.

Затем человек совершает движения ногами, держась руками за брусья. Когда больному удается держать равновесие в такой позе, можно переходить к ходьбе. Пациент пытается ходить, совершая движения руками, двигаясь спиной или боком.
Больным с глазодвигательными нарушениями полезна гимнастика для органа зрения. Пациент пробует фиксировать свой взгляд на определенном предмете или точке, при этом меняя положение головы и тела.
Инвалидность при мозжечковой атаксии
При атаксии Пьера-Мари человек частично или полностью теряет трудоспособность. Поэтому он вправе получить группу инвалидности.
Больной не в состоянии выполнять многие виды работ. Из-за атаксии человек не может длительное время находиться на ногах. Нарушение координации рук препятствует работе, требующей точности движений.
Проблемы с речью мешают деятельности, связанной с общением (например, с клиентами). Если болезнь осложнена интеллектуальными нарушениями, то человек не может заниматься умственным трудом.
Симптоматика заболевания накладывает большие ограничения на работу пациента.
Степень инвалидности устанавливается индивидуально, в зависимости от состояния больного.

Прогноз заболевания
Атаксия Пьера-Мари – это прогрессирующее заболевание. С течением времени симптомы нарастают. Можно сказать, что у этого недуга неблагоприятный прогноз.
Болезнь не влияет на продолжительность жизни и не приводит к летальному исходу. Но изменения в мозжечке необратимы, и со временем пациент теряет трудоспособность.
На сегодняшний день медицина не может вылечить это заболевание, как и многие другие генетические патологии. Однако врачебная помощь поможет несколько замедлить развитие недуга.
Наследственная мозжечковая атаксия Пьера-Мари
Узнать больше о нервных заболеваниях на букву «Н»: Нарушение сна; Нарколепсия; Наследственная мозжечковая атаксия Пьера-Мари; Нарушения спинномозгового кровообращения; Невралгия тройничного нерва; Невралгия подчелюстного и подъязычного узлов; Невралгия языкоглоточного узла; Невралгия ушного узла; Неврастения; Невральная амиотрофия Шарко-Мари-Тута; Невринома слухового нерва; Невринома; Неврит глотки; Неврит лицевого нерва; Неврит; Невроз навязчивых состояний; Невроз глотки; Неврозы; Неврозоподобное заикание; Невропатия бедренного нерва.
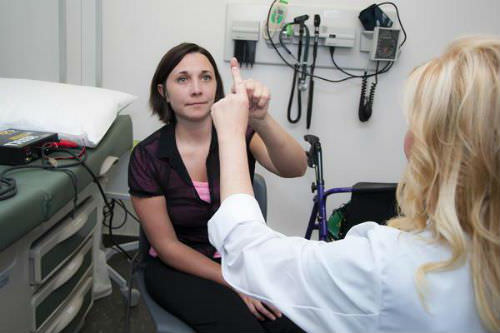
Диагноз устанавливается на базе результатов неврологического и офтальмологического освидетельствования, МРТ головного мозга, УЗДГ или МРА мозговых сосудов, после консультации специалиста — генетика.
Лечение является симптоматическим, включает назначение успокоительных средств, антидепрессантов, миорелаксантов и ноотропов. Абсолютно вылечить заболевание невозможно.
Пациентам рекомендована лечебная физкультура, гидротерапия, витамины.
Общая информация
Впервые заболевание очень детально изложил Пьер Мари в 1893г. Ученый выделил заболевание в особенную форму и определил различные от атаксии Фридрейха проявления. Атаксии Пьера-Мари присущи другие отличительные клинические симптомы, иной способ наследования, более старший возраст проявления.
Заболевание обнаруживается у людей 20-45 лет, независимо от пола. Наблюдается 1 случай болезни на 200 000 человек. Патологические трансформации в структуре при мозжечковой атаксии располагаются в веществе ядер и коры мозжечка. Спинномозговые пути и боковые ответвления спинного мозга выражены не ярко.
Дегенеративные преобразования могут поражать ядра моста продолговатого мозга.
Этиология и течение заболевания
Наследственная мозжечковая атаксия воспроизводится по наследству аутосомно-доминантным способом, развивается при получении патологического гена от одного из родителей.
Внешние причины оказывают содействие развитию заболевания и значительно отягощают течение.
Причинами могут стать разнообразные инфекции (брюшной тиф, бруцеллез, дизентерия, пиелонефрит, пневмония), различные травмы мозга и костей скелета, интоксикации, беременность.
Симптоматика проявляется безостановочно и прогрессирует неуклонно без выраженных промежутков обострения и затихания.
Атаксия Пьера-Мари может видоизменять клинические проявления под влиянием внешних факторов или после перенесенных инфекций. Изменчивость клиники значительно затрудняет установление заключения.
После инфекционного заболевания возможно наступление резкого обострения, а позже — частичное восстановление, которое ошибочно принимается за улучшение.
Клинические признаки

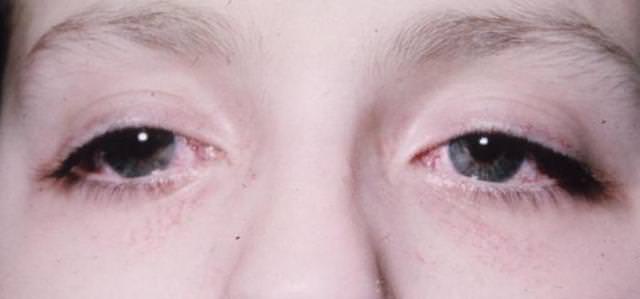
Классические клинические случаи не обходятся без глазодвигательных нарушений и снижения функций зрения. Постепенное омертвение зрительных нервов приводит к сужению полей зрения. Все эти признаки неуклонно прогрессируют. Регистрируется опущение верхнего века. Больной поднимает брови в попытке уменьшить птоз — это придает лицу удивленный вид. Нередко болезнь выражается косоглазием, нарушением бинокулярного зрения, к которым приводит парез отводящего нерва.
К перечисленным нарушениям понемногу присоединяются психические недомогания. Течение заболевания усугубляется снижением умственных процессов и памяти, развивается депрессивное невротическое состояние. При тяжелых клинических случаях бывают эпилептические приступы.
Дифференциальное диагностирование
Заболевание имеет мозжечковый характер и требует неврологической дифференциации от других видов атаксий — сенситивной и вестибулярной. Гипорефлексия, присущая атаксии Фридрейха.
Атаксия Пьера-Мари имеет в неврологической клинике имеет нарастание сухожильных рефлексов, мышечный гипертонус. Имманентны спастические судороги в нижних конечностях, подергивания ступней.
При унаследованной мозжечковой атаксии не обнаруживаются деформации костей скелета, которые присущи болезни Фридрейха.
Клинические выражения рассеянного склероза очень сходны с симптомами наследственной мозжечковой атаксии. Для болезни свойственно неуклонное возрастание клинических проявлений без периодов ремиссии. Трудности постановки диагноза возникают во время или после инфекционных заболеваний, так как меняется характер течения основного недуга.
В таком клиническом случае необходим детальный и точный сбор анамнеза с целью выявления признаков рассеянного склероза — парапареза нижней части тела, сопровождающегося патологическими рефлексами и гиперрефлексией, императивными тазовыми нарушениями, снижением брюшных рефлексов, с височной стороны наблюдается изменение цвета дисков зрительных нервов.
Диагностика
Обязательно проведение компьютерной томографии, магнитно-резонансной томографии, ревизию спинных сосудов, МРА, УЗДГ с целью исключения опухолей мозжечка, вторичных органических патологий, васкулярных расстройств.
Офтальмолог диагностирует зрительные расстройства. Обязательна проверка качества зрения, исследование бинокулярного зрения. Обязательно проведение офтальмоскопии, периметрии, распознавание угла косоглазия.
Желательна консультация врача-генетика для точного определения заболевания.
Лечение и прогноз
Терапия имеет постоянный, комплексный характер. В лечении принимают участие неврологи, офтальмологи и психиатры. Полное излечение от заболевания невозможно. Лечение носит симптоматическую природу.
В основе терапевтических мероприятий лежит назначение антидепрессантов (флуоксетин, тетриндол, амитриптилин, циталопрам), успокоительных средств (настойка пиона, валериана, магния сульфат).
Обязательно применять медпрепараты для стимулирования умственного функционирования и высших психических функций — пирацетам, гамма-аминомасляная кислота, медпрепараты гинго-билоба. Кроме этого назначаются средства, снижающие тонус скелетной мускулатуры — баклофен, мелликтин, кондельфин.

Комплексное лечение оптимизирует общее состояние больного и дает возможность сохранить нормальную жизнь в течение долгого времени, насколько это представляется возможным, когда речь идет об унаследованной мозжечковой атаксии.
Прогноз выздоровления при наследственной мозжечковой атаксии крайне неблагополучный. На протяжении заболевания симптомы обостряются и неизменно приводят к инвалидизации.
Тяжелое протекание болезни приводит к тому, что больной становится прикованным к постели. Смерть наступает от миокардита или сердечной недостаточности.
В лучшем варианте при хорошем присмотре больной может дожить до 40-50 лет.
Наследственная мозжечковая атаксия Пьера-Мари
Наследственная мозжечковая атаксия Пьера-Мари — генетически детерминированное неуклонно прогрессирующее поражение мозжечка, связанное с его дегенеративными изменениями. Развивается после 20 лет. В клинической картине мозжечковая атаксия сочетается с гиперрефлексией, офтальмологическими расстройствами и снижением интеллекта. Диагностический алгоритм предусматривает неврологический и офтальмологический осмотр, МРТ головного мозга, УЗДГ или МРА церебральных сосудов, генетическое консультирование. Радикальная терапия не разработана, осуществляется симптоматическое лечение антидепрессантами, миорелаксантами, седативными и ноотропами. Рекомендуется ЛФК, витаминотерапия и водолечение.
Наследственная мозжечковая атаксия была подробно описана Пьером Мари в 1893г. как отличающаяся от известной на то время атаксии Фридрейха нозологическая форма.
Действительно, атаксия Пьера-Мари имеет другой тип наследования, более старший возраст манифестации и свои клинические отличия.
В связи с этим, несмотря на некоторую общность морфологического субстрата этих заболеваний в виде дегенерации тканей мозжечка и его проводящих путей, в современной неврологии окончательно утвердилось их выделение в самостоятельные нозологические единицы.
Как правило, атаксия Пьера-Мари манифестирует в возрасте от 20 до 45 лет. Мужчины и женщины заболевают одинаково часто. Распространенность патологии составляет 1 случай на 200 тыс. человек.
Морфологические изменения при наследственной мозжечковой атаксии преобладают в тканях ядер и коры мозжечка, менее выражено поражение спиноцеребеллярных путей и боковых канатиков спинного мозга, процессы дегенерации могут наблюдаться в ядрах моста и продолговатого мозга.
Наследственная мозжечковая атаксия Пьера-Мари
Наследственная мозжечковая атаксия имеет доминантный механизм наследования, т. е. развивается при получении дефектного гена от одного из родителей. Зачастую экзогенные причины выступают в роли триггеров, провоцирующих начало заболевания и усугубляющих его течение.
К подобным факторам относятся: различные инфекции (брюшной тиф, сыпной тиф, бруцеллез, сальмонеллез, дизентерия, иерсиниоз, бактериальная пневмония, пиелонефрит и др.), беременность, травмы (ЧМТ, повреждения грудной клетки, переломы таза, сильные ожоги), интоксикации.
Атаксия Пьера-Мари характеризуется постоянным и неуклонно прогрессирующим нарастанием патологической симптоматики. Выделение периодов обострения заболевания и его ремиссии не наблюдается.
Под влиянием перенесенных инфекционных заболеваний и прочих экзогенных воздействий атаксия Пьера-Мари может изменить характер своего течения, что значительно затрудняет ее диагностику.
Так, после перенесенной инфекции наблюдается резкое ухудшение состояния пациента с последующим частичным восстановлением, имитирующим улучшение болезни.
Ведущим симптомокомплексом заболевания является мозжечковая атаксия.
Она включает нарушения походки с отклонением тела в стороны, расстройство статики (в позе Ромберга наблюдается падение в сторону или назад), дискоординацию движений (гиперметрию, дисдиадохокинез, промахивание при выполнении пальце-носовой пробы), размашистую макрографию, дизартрию с прерывистой и замедленной речью, интенционный тремор. Манифестация симптомов обычно происходит с легких нарушений походки, иногда первыми проявлениями становятся боли в пояснице и ногах, по описанию самих пациентов имеющие «стреляющий» характер. Затем возникает атаксия в руках, зачастую сопровождающаяся тремором. В конечностях, лице и на туловище могут наблюдаться непроизвольные мышечные подергивания. Во многих случаях наблюдается снижение мышечной силы. Чувствительность, как правило, сохранена.
Типичные случаи заболевания сопровождаются расстройством зрения и глазодвигательными нарушениями. Первые включают сужение зрительных полей и падение остроты зрения, обусловленные постепенно прогрессирующей атрофией зрительного нерва.
Вторые представлены неполным птозом, недостаточность конвергенции, косоглазием из-за пареза отводящего нерва. Возможны нистагмоидные подергивания глаз. Пытаясь уменьшить явление птоза, пациенты поднимают брови, что придает их лицу удивленное выражение.
Дополняют клинику расстройства психики, снижение мыслительных функций и памяти, развитие депрессивного невроза.
Неврологическое обследование позволяет исключить прочие виды атаксии (вестибулярную, сенситивную) и установить ее мозжечковый характер.
В отличие от атаксии Фридрейха, характеризующейся гипорефлексией и снижением мышечного тонуса, в неврологическом статусе больных при атаксии Пьера-Мари отмечается повышение сухожильных рефлексов и мышечная гипертония.
Особенно типичен спастический тонус в ногах, вызывается клонус стоп. Обычные для болезни Фридрейха выраженные деформации скелета отсутствуют.
Весьма сходной может быть симптоматика наследственной мозжечковой атаксии и рассеянного склероза. Отличительной особенностью первой является постепенное неуклонное прогрессирование без периодов ремиссии, однако различные инфекционные заболевания и травмы могут изменять характер ее течения, вызывая значительные затруднения в постановке диагноза.
В подобных случаях важно тщательное исследование анамнестических данных, выявление типичного для рассеянного склероза симптомокомплекса: более четкой пирамидной симптоматики (обычно нижний парапарез спастического типа со значительной гиперрефлексией и патологическими рефлексами) исчезновения брюшных рефлексов, тазовых нарушений (императивные позывы), побледнения дисков зрительных нервов с височной стороны.
При наличии типичной клиники и прослеживании ее в нескольких поколениях диагноз не представляет для невролога особых затруднений. Спорадические случаи заболевания требуют более углубленного обследования пациента и тщательной дифдиагностики с другими видами мозжечковой атаксии, рассеянным склерозом, нейросифилисом.
При необходимости проводят исключение приобретенной органической патологии: опухолей мозжечка (медуллобластомы, астроцитомы, гемангиобластомы), церебеллита, сосудистых нарушений, сдавлений мозжечка при гематомах или абсцессах головного мозга, окклюзионной гидроцефалии. Для этого используют КТ и МРТ головного мозга, УЗДГ и МРА церебральных сосудов.
Диагностику офтальмологических расстройств проводит офтальмолог. Обследование включает проверку остроты зрения, исследование конвергенции, офтальмоскопию, периметрию, измерение угла косоглазия и пр. Для более точной верификации диагноза может потребоваться консультация генетика.
В лечении пациентов наряду с неврологами принимают участие офтальмологи и психиатры. Поскольку этиотропная терапия пока не разработана, применяется симптоматическое лечение.
В основном это антидепрессанты (амитриптилин, флуоксетин, циталопрам, тетриндол), седативные средства (валериана, пион, магния сульфат), ноотропы (гамма-аминомасляная кислота, пирацетам, экстракты гинго-билоба), препараты, уменьшающие мышечный тонус (меликтин, баклофен, кондельфин). Рекомендованы витамины гр. В, вит. РР и вит.
С; бальнеотерапия, лечебная физкультура. Не маловажное значение имеет соблюдение правильного рабочего режима, исключение физических и психологических перегрузок.
Прогноз относительно выздоровления неблагоприятный. Симптомы заболевания постоянно усугубляются и приводят к инвалидизации. Однако систематическое применение симптоматического лечения, регулярное выполнение специального комплекса ЛФК и других рекомендаций делают благоприятным прогноз для жизни пациента.
Наследственная мозжечковая атаксия пьера мари
Наследственная мозжечковая атаксия — хроническое прогрессирующее заболевание, основным проявлением которого служит мозжечковая атаксия. Заболевание наследственное, передается по аутосомно-доминантному типу. Патологический ген обладает высокой пенетрантностью, пропуски поколений редки.
Основной патологоанатомический признак болезни — гипоплазия мозжечка, в некоторых случаях — атрофия нижних олив, моста мозга (варолиева моста). Наряду с этим, как правило, имеет место комбинированная дегенерация спинальных систем, напоминающая картину спиноцеребеллярной атаксии Фридрейха.
Клиническая картина.
Основной признак болезни — атаксия, которая носит такой же характер, как приатаксии Фридрейха. Заболевание обычно начинается с нарушения походки, к которому затем присоединяется атаксия в руках, нарушение речи, мимики. Выражены статическая атаксия, дисметрия, адиадохокинез.
Могут наблюдаться стреляющая боль в ногах и в области поясницы, непроизвольные мышечные вздрагивания. Значительно снижена сила в мышцах конечностей, отмечается спастическое повышение мышечного тонуса главным образом в ногах. Сухожильные рефлексы повышены, могут вызываться патологические рефлексы.
Часто развиваются глазодвигательные нарушения — птоз, парез отводящего нерва, недостаточность конвергенции; в ряде случаев наблюдались атрофия зрительных нервов, симптом Аргайла Робертсона, сужение полей зрения и снижение остроты зрения. Чувствительные расстройства, как правило, не выявляются.
Один из характерных признаков мозжечковой атаксии — изменения психики, проявляющиеся в снижении интеллекта, иногда депрессивных состояниях.
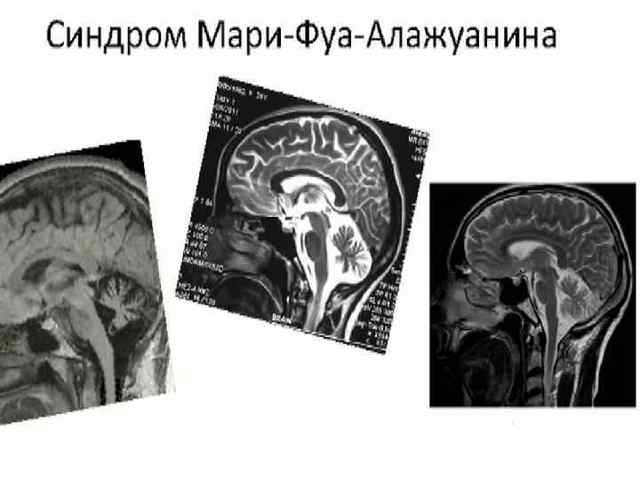
При компьютерной томографии выявляется атрофия мозжечка и ствола головного мозга.
Заболеванию свойственна большая вариабельность клинической картины как в разных семьях, так и внутри одной семьи.
Во многих семьях встречаются рудиментарные формы болезни, иногда экстрапирамидные симптомы. Описаны также многочисленные переходные формы между мозжечковой атаксией и атаксией Фридрейха.
Средний возраст начала болезни — 34 года, в отдельных семьях — более раннее начало в последующих поколениях. Течение заболевания неуклонно прогрессирующее. Как и при атаксии Фридрейха, различные инфекции и другие экзогенные вредные факторы неблагоприятно влияют на проявление и течение заболевания.
Дифференциальный диагноз.
Дифференциальный диагноз между мозжечковой атаксией и атаксией Фридрейха весьма труден. Главными отличительными признаками между этими заболеваниями считаются характер наследования (доминантный при мозжечковой и рецессивный — при атаксии Фридрейха) и состояние сухожильных рефлексов, которые отсутствуют или понижены при атаксии Фридрейха и повышены при мозжечковой атаксии.
Кроме того, при мозжечковой атаксии начало болезни более позднее, редко встречаются костные деформации и расстройства чувствительности, свойственные атаксии Фридрейха, и значительно чаще наблюдаются слабоумие и глазодвигательные нарушения.
Немалые трудности могут возникнуть и при отграничении мозжечковой атаксии от рассеянного склероза, для которого также характерно сочетание мозжечковых, пирамидных и глазодвигательных расстройств. Рассеянный склероз отличается ремиттирующим течением, большей выраженностью нижнего спастического парапареза, тазовыми нарушениями и побледнением височных половин дисков зрительных нервов.
Лечение симптоматическое.
- Применяют специальную систему лечебной гимнастики, направленную преимущественно на уменьшение координаторных нарушений. При назначении упражнений необходимо учитывать возможность сердечной патологии, при наличии которой назначают соответствующую терапию.
- Показаны общеукрепляющие средства (витамины),
- Препараты, влияющие на тканевый обмен (пирацетам, аминалон, ацефен, церебролизин), лечение которыми следует периодически повторять.
Наследственная мозжечковая атаксия Пьера-Мари – что это за заболевание — клиника «Добробут»
Атаксией называется нарушение координации движений при полной сохранности силы мышц. В большинстве случаев заболевание является симптомом, развивающимся на фоне других патологий нервной системы.
Причиной вестибулярной атаксии может стать поражение расположенных в продолговатом мозге вестибулярных ядер, поражение вестибулярного нерва или воспалительный процесс во внутреннем ухе.
Основными проявлениями заболевания является расстройство координации движений и нарушение равновесия.
Виды атаксии:
- вестибулярная (характерные проявления – нарушение походки, сильное головокружение, рвота, тошнота);
- корковая (проявляется нарушением координации и обоняния, изменением психики);
- сенситивная;
- мозжечковая (клинические проявления – нарушение походки, мимики и речи).
Основное лечение сенситивной атаксии направлено на искоренение причины, вызвавшей патологию.
Причины возникновения болезни
Причины атаксии очень разнообразны – от токсического воздействия некоторых фармпрепаратов до воспалительных заболеваний и опухолевидных процессов.
Основные причины:
- заболевания головного мозга (острое нарушение мозгового кровообращения, черепно-мозговые травмы, демиелинизирующие заболевания, гидроцефалия, опухоли мозжечка и мозга);
- заболевания вестибулярного аппарата (невринома вестибулярного нерва, лабиринт, вестибулярный нейронит);
- наследственные заболевания;
- недостаток в организме витамина В12;
- отравление сильнодействующими препаратами, снотворными.
Наследственная мозжечковая атаксия Пьера-Мари
Клиническими проявлениями мозжечковой атаксии являются: утрата плавности речи, гипотония мышц, тремор, окуломоторная дисфункция. Для такой формы заболевания характерна походка с широко расставленными ногами и раскачиванием из стороны в сторону. Вызвать заболевание может сильный авитаминоз, интоксикация, злокачественные опухоли.
Формы наследственной мозжечковой атаксия Пьера-Мари:
- статико-локомоторная (раскачивание при ходьбе, шаткость походки);
- статическая (нарушение координации движений при стоянии);
- кинетическая (проявляется нарушением координации при выполнении точных движений);
- сенситивная (нарушение координации при выполнении движений с закрытыми глазами);
- вестибулярная (нарушение координации, сопровождающееся тошнотой, рвотой, нистагмом);
- корковая.
Диагноз «поздняя мозжечковая атаксия» мужчинам ставится в два раза чаще, нежели женщинам. К этой группе относятся идиопатические мозжечковые дегенерации и аутосомно-доминантные мозжечковые атаксии.
Спиноцеребеллярная атаксия у ребенка
Спиноцеребеллярной атаксией называется нейродегенеративная патология, объединяющая несколько типов аутосомно-доминантных заболеваний, каждое из которых рассматривается отдельно.
Особенностью патологии является быстрое нарастание клинических проявлений. К симптомам атаксии относятся: неловкость движений и шаткость при походке, тремор рук, невозможность выполнять точные движения.
При прогрессировании болезни возникают проблемы с мочеиспусканием, наступает атрофия мышц, паралич.
На нашем сайте https://www.dobrobut.com/ вы найдете больше информации по теме «спиноцеребеллярная атаксия у ребенка» и сможете записаться на консультацию к врачу. Прием ведут специалисты высшей категории.
Симптомы атаксии Фридрейха
Атаксия Фридрейха – генетическое заболевание, характеризующееся поражением нервной системы с экстраневральными нарушениями в работе сердца, эндокринной системы и опорно-двигательного аппарата. Патология начинается в молодом возрасте с неврологических проявлений.
Основными методами диагностики являются МРТ (магнитно-резонансная томография) и нейрофизиологическое тестирование. Могут быть назначены дополнительные обследования – исследования гормонального фона, анализ крови на сахар, биохимия, рентген позвоночника, УЗИ сердца.
Также потребуется консультация кардиолога, офтальмолога и эндокринолога.
Симптомы атаксии Фридрейха: неуверенность и шаткость при ходьбе, нарушение координации рук, тремор, изменение почерка, слабость в ногах, нарушение речи, снижение слуха.
Диагностика заболевания
После тщательного осмотра и сбора анамнеза врач обязательно назначит дополнительные обследования.
Диагностика при атаксии конечностей включает в себя:
- электроэнцефалографию (ЭЭГ);
- магнитно-резонансную томографию (МРТ) головы и спинного мозга;
- компьютерную томографию (КТ);
- магниторезонансную ангиографию (МРА);
- лабораторный анализ крови, токсикологический анализ;
- ДНК-тест, если подозревается синдром Луи Бар (атаксия телеангиэктазия);
- неврологический осмотр;
- консультацию кардиолога, отоларинголога, нейрохирурга.
Лечение атаксии
Основное лечение направлено на устранение причины, вызвавшей заболевание. Курс состоит из ангиопротекторов, ноотропов, антибиотиков (при необходимости), гормонов и витаминов группы В. Отличным дополнением станет гимнастика и ЛФК при синдроме атаксии.
- При своевременном и квалифицированном лечении прогноз приобретенного заболевания благоприятен, чего не скажешь о наследственных атаксиях.
- Связанные услуги:
Компьютерная томография
Магнитно-резонансная томография
19.6. Наследственная мозжечковая атаксия Пьера Мари
В 1893
году Пьер Мари выступил с предложением
разбить группу семейных атаксий на две
болезни: болезнь Фридрейха и половую
форму, которую он назвал наследственной
мозжечковой атаксией.
По
сравнению с болезнью Фридрейха данная
болезнь отличается поздним началом
(30-40 лет), отсутствием костных деформаций
и склонностью к развитию церебральных
симптомов (дисфагия, слабоумие) и в
частности глазных (птоз, паралич
отводящего нерва, затруднение
конвергенции, симптом Аргайлла-Робертсона,
атрофия зрительных нервов).
Мозжечковая
атаксия Пьера Мари характеризуется
аутосомно-доминантным типом наследования.
Патологический ген локализован в области
6р23. В последние годы большое значение
в патогенезе наследственных атаксий
придается нарушениям в системе
нейромедиаторных аминокислот.
Найдено
снижение содержания глютамата, аспартата,
ГАМК и дисбаланс других аминокислот в
различных отделах головного и спинного
мозга при разных формах наследственных
атаксий.
Найденные изменения не являются
первичными метаболическими дефектами
и представляют собой лишь звенья сложного
комплекса биохимических нарушений,
ведущих к дегенерации нейронов ЦНС.
Атаксия
Пьера Мари начинается постепенно с
расстройства походки. Нередко
отмечается атаксия речи, спастические
парезы ног, повышение сухожильных
рефлексов. Часто развивается депрессия
и страдает память.
В
настоящее время болезнь Пьера Мари не
считается единой нозологической
единицей. По особенностям клинического
течения выделяют разные ее варианты,
многие из которых включены в группу
наследственных оливопонтоцеребеллярных
атаксий.
В
отличие от последних при атаксиии Пьера
Мари нет экстрапирамидных нарушений,
гиперкинетического или амиостатического
синдромов, нет грубых парезов конечностей.
Патолого-анатомически
при
атаксии Пьера Мари выявляется гипоплазия
мозжечка с отсутствием клеток Пуркинье,
резкая атрофия моста. Дегенеративные
изменения отмечают в пирамидных и
спиномозжечковых путях.
КТ и МРТ выявляют
четкое уменьшение в объеме мозжечка,
резкую выраженность его борозд.
Эффективного
лечения
в
связи с отсутствием знаний о первичных
белковых продуктах мутантных генов,
при наследственных атаксиях до
настоящего времени нет.
Комплексное
лечение включает в себя применение
витаминов
группы В, церебролизина, ноотропов,
эссенциале, витамина Е, АТФ, рибоксина,
аденила. Регулярно
должна проводиться лечебная
физкультура. Курсы комплексного лечения
позволяют замедлить прогрессирование
болезни и имеют большое психологическое
значение.
19.7. Болезнь Фридрейха
В 1862
году Фридрейх описал болезнь, получившую
впоследствии его имя, и характеризующуюся
дегенерацией задних и боковых столбов
спинного мозга (мозжечковые и пирамидные
пути), дегенерацией клеток Пуркинье
и зубчатого ядра мозжечка, дегенерацией
задних корешков.
Частота
болезни 2-7 человек на 100 000 населения.
Это аутосомно-рецессивное заболевание
с частотой гетерозиготного носительства
около 1%.
Начало
болезни приходится на 1-2-е десятилетие
жизни с большей частотой ее появления
в период полового созревания. Начинается
болезнь с поражения задних столбов
поясничного отдела спинного мозга,
и процесс распространяется книзу и
кверху, не захватывая продолговатый
мозг. Иногда встречается поражение
зрительного и слухового нервов.
Клинически
болезнь
выражается сочетанием смешанной атаксии,
повышением мышечного тонуса по пирамидному
типу и снижением глубоких сухожильных
рефлексов, нистагмом. Кроме того, ген
семейной атаксии связан весьма
прочной коррелятивной связью с
изменениями со стороны скелета
(кифосколиоз, стопа Фридрейха) и сердечной
мышцы.
Походка
больных становится атактической
(сенсорно-мозжечковой) за счет
поражения задних столбов и мозжечка.
При исследовании координации можно
найти дисметрию, адиадохокинез, изменение
почерка, характерно пошатывание в
темноте. Интеллект обычно сохранен,
хотя встречается сочетание болезни
Фридрейха с дебильностью.
Мозжечковая
и сенситивная атаксии постепенно
нарастают, появляется слабость и
атрофия мышц ног, а позже данная
симптоматика распространяется и на
руки. Может развиться атрофия зрительных
нервов. Более чем у 90% больных наблюдается
поражение сердца (кардиомиопатия),
бывают и эндокринные расстройства
(диабет, инфантилизм, дисфункция
яичников).
Болезнь
медленно, но неуклонно прогрессирует,
не дает ремиссий. Биохимические
причины болезни не выяснены. КТ
малоинформативна, на МРТ можно увидеть
атрофию спинного мозга.
Вариабельность
болезни можно объяснить существованием
в одном гене нескольких мутаций,
имеющих разное клиническое проявление.
Было установлено, что дефект имеется в
9-й хромосоме, в области 9 ql3-q21.1.
Поскольку сам ген пока неизвестен,
возможна косвенная ДНК-диагностика
заболевания, а также пренатальная
диагностика болезни и диагностика
гетерозиготного носительства мутантного
гена.
Лечение
симптоматическое.
Важное место занимает тренировка систем,
участвующих в механизме статокинетики,
массаж нижних конечностей, ортопедические
мероприятия. Из медикаментозных средств
применяют витамины
группы В, витамин Е, церебролизин, АТФ,
кокарбоксилазу, сердечные средства (по
показаниям), антихолинэстеразные
средства.
Comments
(0 Comments)