Талассемия, болезнь Кули, мишеневидно-клеточная гемолитическая анемия
Что же за болезнь талассемия Это наследственная гемолитическая анемия.
Заболевание, как видно из определения, чаще всего передаётся по наследству от родителей. Возникает оно из-за ошибки (мутации) в генах, кодирующих в нашем организме структуру молекулы гемоглобина. У людей, которые обладают таким «поломанным» геном, гемоглобин вырабатывается с изменёнными свойствами. Эритроциты (красные клетки крови) из-за этого становятся очень маленькими и бледными, если рассмотреть их в микроскоп. Кроме того, эти эритроциты по сравнению с нормальными очень легко разрушаются. От этого у людей с тяжелой и среднетяжелой формами талассемии периодически резко снижается уровень гемоглобина и эритроцитов в крови и появляется желтуха (это состояние называется гемолиз — внезапное разрушение эритроцитов). Из разрушенных эритроцитов в кровь попадает много свободного гемоглобина, который печень быстро перерабатывает в билирубин. А билирубин красит кожные покровы и слизистые оболочки, включая белки глаз, в ярко жёлтый цвет (это и есть желтуха, похожая на ту, что бывает у новорожденных).
Помочь ребенку с внезапным гемолизом можно с помощью переливания донорской крови. Но лишь временно, до следующего гемолиза (разрушения эритроцитов), который часто возникает в ответ на вирусную инфекцию или другой стресс. А иногда просто без видимых причин.
Радикальным методом лечения талассемии в самых тяжелых случаях может стать трансплантация костного мозга.
Но тяжёлая или иначе большая форма талассемии в России встречается, к счастью, редко! Гораздо чаще детские гематологи ставят диагноз малой формы талассемии, а она почти не имеет симптомов у большинства наших маленьких пациентов. Поэтому и диагноз правильный врачу-педиатру поставить сложно, как я уже говорила, поначалу малую форму талассемии легко можно спутать с железодефицитной анемией
Поэтому мы и уделим ей сегодня особое внимание
От чего же зависит разница в симптомах талассемии?
В каждой вашей клетке по 46 хромосом, а точнее 23 пары хромосом. В каждой паре одна хромосома пришла к вам от мамы, другая от папы. Есть правда исключительные клетки у каждой мамы — это яйцеклетки в яичниках, они содержат не 46, а всего 23 хромосомы. У отцов наших детей тоже есть такие исключения — это сперматозоиды, в которых так же по 23 хромосомы. Когда в момент оплодотворения ядра сперматозоида и яйцеклетки сливаются, каждая из 23 хромосом мамы находит соответствующую из 23 хромосом папы, и они образую те самые пары. Получается в каждой клетке вашего будущего малыша снова по 46 хромосом.
Зачем все так сложно устроено? Причин у природы было много, и одна из них — наша с вами безопасность.
В каждой из парных хромосом, в общем-то, одни и те же гены. Но если у кого-то из родителей на месте одного из этих генов случайно окажется ошибка (мутация), то ген, полученный от второго родителя, подстрахует, и организм сможет работать в нормальном режиме.
Симптомы талассемии
Признаки большой (гомозиготной) b-талассемии проявляются уже в течение 1-2-го года жизни ребенка. Больные дети имеют характерное монголоидное лицо, седловидную переносицу, башенный (четырехугольный) череп, гипертрофию верхней челюсти, нарушение прикуса, гепато- и спленомегалию. Проявлениями анемизации служат бледный или землисто-желтушный цвет кожных покровов. Поражение трубчатых костей сопровождается отставанием в росте и патологическими переломами. Возможно развитие синовита крупных суставов, калькулезного холецистита, язв нижних конечностей. Фактором, осложняющим течение b-талассемии, выступает гемосидероз внутренних органов, приводящий к развитию цирроза печени, фиброза поджелудочной железы и, как следствие, — сахарного диабета; кардиосклероза и сердечной недостаточности. Больные восприимчивы к инфекционным заболеваниям (кишечным инфекциям, ОРВИ и др.), возможно развитие тяжелых форм пневмонии и сепсиса.
Малая (гетерозиготная) b-талассемия может протекать бессимптомно или с минимальными клиническими проявлениями (умеренным увеличением селезенки, незначительно выраженной гипохромной анемией, жалобами на повышенную утомляемость). Аналогичная симптоматика сопровождает течение гетерозиготной формы a-талассемии.
При гомозиготной форме a-талассемии альфа-цепи полностью отсутствуют; фетальный гемоглобин у плода не синтезируется. Данная форма талассемии несовместима с жизнью, что приводит к внутриутробной гибели плода вследствие развивающегося синдрома водянки или самопроизвольному прерыванию беременности. Течение гемоглобинопатии Н характеризуется развитием гемолитической анемии, спленомегалии, тяжелых костных изменений.
Основные способы лечения
Болезнь талассемия подразумевает комплексное лечение, дающее возможность предотвратить дальнейшее ее прогрессирование. В данном случае лечение талассемии имеет свои определенные особенности. Обнаружение данной кислородной нехватки на клеточном уровне свидетельствует о том, что пациенту необходимо экстренное переливание крови. На начальных этапах лечения заболевания данному пациенту требуется порядка 10 переливаний. Такой способ крайне необходим для восстановления уровня гемоглобина в крови до нормальных показателей. В последующем общее число переливаний уменьшается, но уровень гемоглобина тщательно контролируется.
Данная методика лечения талассемии направлена на:
- улучшение общего состояния пациента;
- устранение возможного пагубного влияния заболевания на костную систему;
- повышение и укрепление иммунной системы;
- предотвращение дальнейшего увеличения селезенки;
- благоприятное развитие ребенка.
Особенно важно для специалистов во время лечения вывести излишки железа из организма. В данном случае назначаются внутримышечные инъекции Десферал, особенно в сочетании с витамином С
Нередко для стойкого улучшения общего состояния здоровья пациенту может быть показано проведение оперативного лечения с целью трансплантации костного мозга.
К наиболее распространенным и эффективным методам лечения Талассемии относятся:
Переливание эритроцитов. Особенно актуально при диагностировании тяжелой формы заболевания. Со временем у пациента начинает развиваться так называемая трансфузионная зависимость. Такая реакция организма характеризуется стабильным снижением гемоглобина. Это может угрожать жизни пациента или заболевание начнет активно прогрессировать. Такая процедура имеет свои существенные недостатки. В данном случае проблемы могут возникнуть с поиском наиболее подходящего донора, так как требуется тщательный индивидуальный подбор.
Удаление селезенки. Данная процедура имеет название спленэктомия, при этом метод считается необходимым из-за влияния увеличенного органа на показатели гемоглобина. Таким образом, устранение данного внутреннего органа может повлиять на течение заболевания. Операция может проводиться только при достижении 8-летнего возраста. Однако и здесь есть свои недостатки, так как может быть спровоцировано увеличение печени
Следует с особой осторожностью принимать решении об удалении органа.
Трансплантация костного мозга. Считается одним из радикальных методов терапии
Важно проведение биологического исследования всех членов семьи на совместимость
Это даст возможность найти более подходящего донора
К тому же данная процедура имеет сравнительно высокую стоимость.
Не меньшей популярностью пользуется при устранении заболевания талассемия и лечение с помощью генной инженерии
В данном случае важно придерживаться специально подобранной диеты (стол №5), употреблять напитки в составе которых присутствует танин, например, чай или какао. Для поддержания функциональности печени пациенту могут быть назначены медикаментозные средства — гепатопротекторы: липоевая кислота, витамин Е, Эссенциале, а также курс желчегонных трав — мяты, барбариса и тюбажи
Существуют также способы лечения других форм заболевания талассемиия, например, промежуточной. В данном случае пациенту не требуется постоянное переливание крови, можно ограничиться лишь единичными процедурами, которые проводятся в промежутке от нескольких недель до нескольких месяцев.
Подводя итог всему вышесказанному, следует отметить, что такая болезнь как талассемия лишь в единичных случаях может быть полностью устранена, при условии, что оно будет диагностировано на ранних стадиях развития
Важно проводить необходимое лечение с целью предотвращения возможных осложнений и продления жизни пациента
Диагностические мероприятия
Диагностика заболевания у пациентов осуществляется путем проведения определенных исследований, к основным из которых относятся:
- изучение костного мозга;
- электрофорез гемоглобина;
- сдача анализа крови на выявление уровня гемоглобина.
В большинстве случаев при талассемии достаточно пациенту сдать общий анализ крови, результаты которого уже могут свидетельствовать о присутствии патологического процесса и нарушении состояния здоровья ребенка.
Комплексное обследование до рождения ребенка следует проводить на 11 недели беременности. Для этого берется фрагмент плаценты, а зараженность плода талассемией определяется посредством изучения околоплодных вод примерно на 16 неделе.
В настоящее время особой популярностью пользуется новейшая технология предимплантантной генетической диагностики талассемии. Такой способ дает возможность матери в дальнейшем родить здорового ребенка
Так как основная диагностика представляет собой лабораторное изучение анализов, то есть смысл обратить внимание на расшифровку полученных результатов и на то, каковы отклонения от нормы основных показателей:
- при изучении крови уровень гемоглобина снижается до распространенных пределов — 50-30 г/л;
- неутешительный цветной показатель в результате расчетов варьируется в пределах 0,5 и ниже;
- эритроциты имеют измененные размеры;
- резистентность кровяных телец явно увеличенная;
- число ретикулоцитов значительно превышает нормальные значения.
Изменен в худшую сторону и билирубин, что в свою очередь говорит о чрезмерном количестве присутствующего железа. Такие нарушения могут спровоцировать возникновение параллельных заболеваний — цирроза печени и развитию сахарного диабета. В отдельных случаях не исключено поражение сердечной функциональности.
Как жить с таким диагнозом прогноз
Переливания эритроцитной массы в сочетании с медикаментозной терапией позволяет улучшить качество жизни больного. Однако такое комплексное лечение не в состоянии полностью излечить пациента, поскольку имеет поддерживающее значение. Больные с тяжёлыми формами талассемии, как правило, живут не более 5–7 лет, при этом находясь в постоянной зависимости от переливаний донорских эритроцитов. Патологическая деформация внутренних органов прогрессирует, доставляя ребёнку боль.
Продолжительность жизни при талассемии средней тяжести 14–20 лет, положительный прогноз лечения врачи дают только в случае удачной пересадки костного мозга.
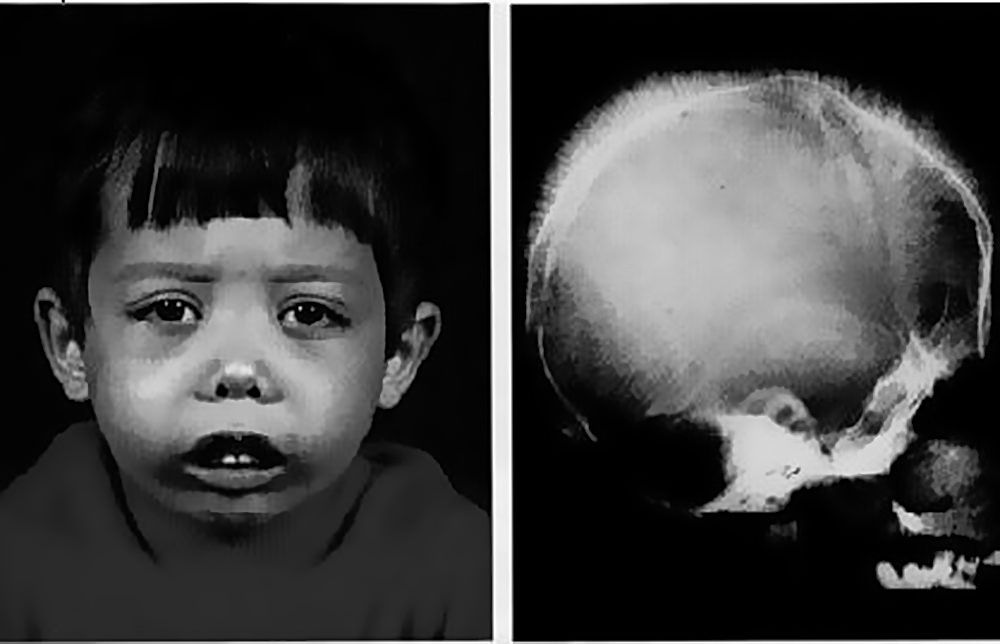
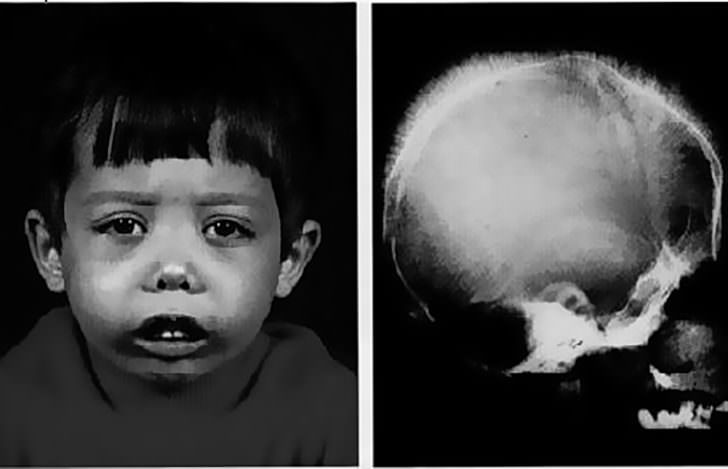 Лёгкая форма талассемии не сказывается на длительности жизни
Лёгкая форма талассемии не сказывается на длительности жизни
Разновидности заболевания при нарушении бета-цепочек
Гены, которые контролируют выработку бета-цепочек, могут находиться в разном состоянии:
- Нормальный здоровый ген. У здоровых людей он находится в норме, что обеспечивает выработку правильных цепочек гемоглобина.
- Ген с частичными отклонениями. Такая патология позволяет организму вырабатывать нормальный гемоглобин, однако, он продуцируется в недостаточном количестве.
- Ген искажен настолько, что полностью препятствует формированию бета-цепей.
 Структура бета-цепей
Структура бета-цепей
В зависимости от масштабов заболевания различают следующие виды бета-талассемии:
- Минорная талассемия. При таком виде заболевания поврежден только один ген. Симптоматика практически отсутствует. Может наблюдаться незначительная анемия. Помимо малокровия, человек не жалуется на состояние своего здоровья.
- При более серьезной патологии гена, отвечающего за синтез бета-цепей, наблюдается талассемия интермедиа. В результате нарушения процесса производства гемоглобина эритроциты формируются либо недоразвитыми, либо маленьких размеров. Переливания крови не являются необходимостью, однако, последующее состояние организма напрямую зависит от его способности жить при низком уровне гемоглобина.
- Бета-талассемию типа «майор» характеризует нарушением всех генов, отвечающих за синтез бета-цепей. Больные таким типом заболевания требуют регулярных переливаний крови. Такая процедура позволяет поддерживать жизнь пациента.
Лечение и прогноз талассемии
Лечебная тактика при различных формах талассемии неодинакова. Так, пациенты с малой b-талассемией в лечении не нуждаются. С другой стороны, больным с гомозиготной b-талассемией с первых месяцев жизни требуется проведение гемотрансфузионной терапии (переливание размороженных или отмытых эритроцитов), введение хелатирующих препаратов, связывающих железо (дефероксамина), глюкокортикоидов при возникновении гемолитических кризов. При всех формах талассемии показан прием препаратов фолиевой кислоты и витаминов группы В.
При гиперспленизме (особенно на фоне гемоглобиноза Н) требуется удаление селезенки (спленэктомия). Из-за склонности к присоединению инфекционных осложнений больным рекомендуется обязательная вакцинация против пневмококковой инфекции. Многообещающим методом лечения талассемии служит трансплантация костного мозга от гистосовместимого донора.
Прогноз больших форм талассемии неблагоприятный; больные погибают в младенческом или молодом возрасте. При гетерозиготной бессимптомной форме талассемии продолжительность и качество жизни в большинстве случаев не страдают. Первичная профилактика талассемии включает предупреждение браков между гетерозиготными носителями генов заболевания, а при высоком генетическом риске рождения больного потомства – отказ от деторождения.
Талассемия – это ряд заболеваний крови, которые могут передаться по наследству ребенку от родителей. При этом заболевании наблюдается нарушение состава гемоглобина крови, а именно функции альфа — или бета — цепей гемоглобина, а это выражается в нехватке кислорода для внутренних органов человека, которые из-за этого не могут нормально функционировать.
Гемоглобин в целом состоит из комплекса клеток содержащих железо, и клеток содержащих в себе белок. Белок в свою очередь содержит в себе пару альфа — цепочек и пару бета – цепочек. Если же происходит нарушение в одной из цепочек, то происходит изменение в составе гемоглобина крови. При таком процессе белок начинает постепенно разрушать эритроциты крови, которые уже не смогут в полной мере питать клетки кислородом, что приводит к анемии и железо начинает накапливаться в тканях и на внутренних органах.
Если же альфа-цепей больше нормы, и они уже являются лишними в структуре гемоглобина, то это состояние является опасным. В костном мозге новые ядерные эритроциты поддаются разрушению, а зрелые красные клетки, которые уже находятся в периферической крови, тоже могут распасться.
Еще одно заболевание крови, которое передается наследственным путем, есть серповидноклеточная анемия. Талассемия и серповидноклеточная анемия являются основными заболеваниями с нарушением состава клеток и составляющих крови. Из самого названия этого заболевания можно понять, что кровяные клетки деформируются и приобретают форму серпа или кольца, а должны иметь ровную форму. Они перестают правильно выполнять свою функцию и могут стать причиной закупорки маленьких кровеносных сосудов и нарушить кровоток в венах. При таком состоянии красные кровяные клетки погибают, вызывая при этом анемию.
Заболевание талассемия берет свое начало из стран средиземноморского бассейна. Там это заболевание называется средиземноморской анемией. Особенно в Африке талассемия довольно частое явление, по причине малярии. Также от талассемии страдает население Индии, Ближнего Востока и Восточной Азии.
Клиническая картина править править код
При талассемии характерны гипохромная анемия, анизоцитоз эритроцитов, наличие мишеневидных форм эритроцитов (пятно гемоглобина в центре клетки, напоминающее мишень). При этом содержание сывороточного железа нормальное или повышенное. Компенсаторная гиперплазия костного мозга ведёт к нарушениям в строении лицевого черепа. Череп может стать квадратным, башенным; нос приобретает седловидную форму; нарушается прикус и расположение зубов. Отмечается желтушность кожи и слизистых оболочек. Селезёнка и печень увеличены. Больные подвержены инфекционным заболеваниям. Рано начавшаяся анемия обуславливает физическое и умственное недоразвитие ребёнка.
Талассемия – наследственные гемоглобинопатии, характеризующиеся угнетением синтеза цепочечных белковых молекул, образующих структуру гемоглобина. Это приводит к повреждению мембраны эритроцитов и разрушению красных клеток крови с развитием гемолитических кризов. Признаками талассемии служат характерные костные изменения, гепатоспленомегалия, анемический синдром. Диагноз талассемии подтверждается клиническими и лабораторными данными (исследованием гемограммы, гемоглобина, миелограммы, электрофоретическим методом). Возможна пренатальная диагностика талассемии. В лечении талассемии применяются гемотрансфузии, терапия десфералом, спленэктомия, трансплантация костного мозга.
Большая талассемия анемия Кули, анемия Средиземноморья
Некотороые образцы электрофоретического гемоглобина имеют характерные особенности.
1. Классификация синдромов бета-талассемии:
- гомозиготная β0: НbА отсутствует; HbF и НbА2 присутствуют;
- гомозиготная β+: НbА, НbА2 и HbF все обнаруживаются;
- HbF — 10 — 90%; НbА снижен; НbА2 может быть в норме, низким или высоким.
2. В анализе крови выраженная гипохромная микроцитарная регенеративная гемолитическая анемия. Обычно уровень гемоглобина от 2,0 до 6,5 г/дл, гематокрит 10 — 24%, число эритроцитов 2 — 3 млн., эритроцитарные индексы понижены.
3. Мазок крови показывает анизоцитоз, пойкилоцитоз, мишеневидные клетки, сфероциты, гипохромные, фрагментированные и причудливой формы эритроциты; также много ядерных эритроцитов — базофильные включения, кольца Кебота, сидероциты.
4. Число ретикулоцитов выше нормы.
5. Число лейкоцитов повышено, лейкоформула в норме или со сдвигом влево. Тромбоциты в норме.
6. Костный мозг при анемии Кули ячеистого строения, выявляются эритроидная гиперплазия и повышение железа.
7. Сывороточное железо и ОЖСС повышены. После 5-летнего возраста железо-связывающая способность сыворотки обычно полная.
Лабораторные данные при анемии Кули показывают гемолиз и дисфункцию печени, например, повышение сывороточной ЛДГ, ACT, АЛТ и непрямого билирубина, появление уробилиногена в моче и кале; сывороточный гаптоглобин и гемопексин (норма 0,5-2 мг/дл) сильно снижены или отсутствуют.
Дисфункция печени обусловливает нарушение синтеза V, VII, IX, XI факторов свертывания, протромбина, изменяются результаты анализов крови на свертываемость. При большой талассемии время выживаемости эритроцитов понижено, а осмотическая хрупкость снижена, механическая хрупкость повышена.
Лабораторные данные при анемии Кули могут свидетельствовать об осложнениях:
- вторичный гиперспленизм — обычно развивается в возрасте 5-10 лет, обнаруживают, когда рекомендованная трансфузия становится более 250 мг/кг веса тела, при этом показана спленэктомия;
- гемосидероз — фиброз печени и цирроз; эндокринопатии с гипофункцией половых, щитовидных желез;
- заболевания костей, например, изменение черепа, рахит, остеопороз);
- сердечные заболевания.
- Протеинурия, гипостенурия, неспособность подкислять мочу, повышение уробилина и уробилиногена с окрашиванием мочи в темный цвет.
Носительство бета-талассемии присутствует у обоих родителей.
Пренатальная диагностика большой талассемии возможна с 16-й недели гестации. В 85% случаев проводят анализ ДНК амниотических клеток; остальные случаи могут быть диагностированы при определении соотношения α:β – цепей в фетальной крови (полученной путем фетоскопии).
Классификация гомозиготная и гетерозиготная альфа- и бета-талассемия
Заболевание может передаваться от одного родителя, то есть гетерозиготно. В таком случае количество мутированных генов минимально, а патология обычно протекает в лёгкой форме. При унаследовании изменённых генов сразу от отца и матери (гомозиготно) талассемия может иметь различные формы: лёгкую, тяжёлую и даже несовместимую с жизнью младенца.
В зависимости от того, какая из полипептидных цепей гемоглобина была подвержена мутации, различают основные формы заболевания:
- Альфа-талассемия характеризуется угнетением синтеза α-цепей, вместо них образуются избыточные β- или γ- цепи. Патология возникает при отсутствии одного или нескольких генов хромосомы. Местоположение гена на участке хромосомы называют локусом. Всего насчитывают 4 локуса, ответственных за синтез альфа-цепей:
- мутации одного участка хромосомы не проявляются клинически, а человек является носителем «спящего» гена;
- при генетических изменениях в двух локусах анемия может протекать в лёгкой форме, поскольку оставшиеся два способны передавать правильную информацию, благодаря чему часть гемоглобина продуцируется без структурных изменений;
- нарушение в трёх локусах сопровождается избыточной выработкой нестабильных бета-цепей, у больных наблюдается микроцитарная гипохромная анемия;
- отсутствие всех четырёх кодирующих элементов несовместимо с жизнью, при такой патологии плод погибает внутриутробно от водянки.
- Бета-талассемия встречается намного чаще. Но поскольку за воспроизводство β-цепей отвечают только два гена, шансы на тяжёлое протекание болезни значительно выше. Выделяют две основных и две промежуточных формы патологии:
- Большая β-талассемия (анемия Кули или майор талассемия) — среднетяжелая форма заболевания, характеризующаяся образованием фетального гемоглобина. Она развивается гомозиготно при унаследовании двух полностью мутированных генов от обоих родителей. Симптомы заболевания проявляются ярко с первых дней жизни.
- Промежуточная β-талассемия (интермедиа) имеет доброкачественное течение, так как её симптомы выражены в меньшей степени и могут проявляться в позднем возрасте. Развивается при унаследовании двух частично мутированных генов (гомозиготно) или одного патологически изменённого гена (гетерозиготно).
- Малая β-талассемия (минор) — при мутации одного из генов заболевание протекает в форме лёгкой гипохромной анемии.
- Минимальная β-талассемия не сопровождается возникновением симптомов.
Форма заболевания и степень тяжести его протекания зависят от того:
- какие гены ребёнок унаследовал от родителей;
- какое количество хромосом повреждено;
- как это отражается на качественном и количественном синтезе глобина.
Выделяют три степени тяжести болезни:
- тяжёлую. Возникает при гомозиготном наследовании генов от двух родителей. Как правило, такие дети погибают в период новорождённости или в течение нескольких лет;
- среднюю. Проявляется анемией и увеличением селезёнки, может возникать при гетерозиготном наследовании при отсутствии двух или трёх генов, ответственных за синтез α-цепей, а также частичном разрушении одного гена при β-талассемии;
- лёгкую. Талассемии диагностируется только при гетерозиготном унаследовании бракованного гена от носителя. Болезнь проявляется в виде хронического анемического синдрома.
Признаки талассемии
Первые признаки талассемии будут появляться еще в периоде новорожденности. Талассемия у детей является крайне жестким и суровым заболеванием. Благодаря тому, что в организме при талассемии запущен процесс тканевой гипоксии — костные структуры ребенка не формируются нормально, происходит нарушение развития и психики.
Так как талассемия сопровождается крупным гемолизом, то кожные покровы будут иметь характерный «гемолитический» цвет
Важно отметить, что такая желтуха будет иметь свой особенный лимонный или «канареечный» цвет, что объясняется наложением желтухи на анемическую бледность
Еще одним возможным признаком талассемии будет объективное пальпируемое увеличение селезенки и печени. Селезенка накапливает в себе распадающиеся эритроциты и при большом их количестве увеличивается, происходит так называемая «рабочая гипертрофия» органа. Увеличение же печени объясняется гемосидерозом и массированным отложением его ферментов в клетках печени. В дальнейшем печень будет, наоборот уменьшаться, и приобретать более плотную консистенцию.
К этим признакам также добавятся типичные общие признаки анемии: слабость, упадок сил, потеря аппетита, сильные головокружения, которые могут сопровождаться обмороками. Еще один яркий признак развивающейся анемии — на фоне полного благополучия при адекватных физических нагрузках пациенты начинают отмечать у себя резкое развитие сильной одышки. Этот процесс также объясняется нарастающей гипоксией организма, которую он пытается устранить, заставляя себя чаще дышать. Но этот признак может быть адекватным только при наличии нормальной функции сердечно-сосудистой и дыхательной систем.
Большая форма талассемии является подвидом талассемии с высоким летальным исходом — такой ребенок может умереть уже на первом году жизни. При длительном лечении и менее активной форме продолжительность жизни более высокая, но такие больные с талассемией страдают от обширных гемосидерозов: появляется гастрит, миокардиодистрофические поражения и поражения суставов.
Альфа-талассемия
Люди, чей гемоглобин не производит в достаточном количестве альфа-белок, страдают от альфа-талассемии.
Существует несколько типов альфа-талассемии:
1. Бессимптомный.
Это состояние, как правило, не вызывает никаких проблем со здоровьем, поскольку уровень отсутствия альфа-белка очень мал. Функция гемоглобина при этом не нарушается. Человек, больной таким заболеванием, называется «молчаливым носителем». У таких людей могут родиться дети с признаками альфа-талассемии.
2. Мутация альфа-гемоглобина.
Наблюдается, в основном, на территории Ямайки, где впервые было обнаружено это состояние крови. Проблем со здоровьем у таких пациентов обычно нет.
3. Мягкая форма альфа-талассемии.
4. Синдром Н-гемоглобина.
В этом состоянии у пациентов наблюдаются серьезные проблемы со здоровьем, например, увеличение селезенки, вирусные инфекции. Эта форма заболевания названа в честь аномального гемоглобина Н, который разрушает красные кровяные клетки.
5. Хронический Н-гемоглобин.
Более опасный для здоровья человека синдром, приводящий к появлению серьезной анемии, проблемам с внутренними органами.
6. Гомозиготный Н-гемоглобин.
Этот симптом наблюдается у детей, чьи родители являются носителями хронической формы Н-гемоглобина.
Еще одна форма альфа-талассемии называется водянкой плода. При этом состоянии у новорожденного отсутствуют в ДНК альфа-гены, которые трансформируют гамма-глобины, произведенные организмом, в гемоглобин Барта.
Большинство детей с таким состоянием умирают до или сразу после рождения. В некоторых, крайне редких случаях, внутриутробное переливание крови позволяют ребенку с водянкой родиться живым, однако таким детям требуется пожизненное переливание крови и медицинская помощь.
Comments
(0 Comments)