Буллезный эпидермолиз: что такое, основные симптомы, 3 вида, способы лечения, видео
Буллезный эпидермолиз
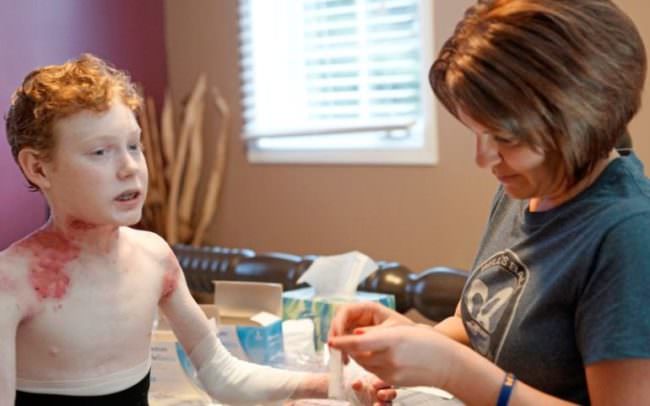
Буллезный эпидермолиз – группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий — «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии.
Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза.
Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие.
Обратите внимание
Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии.
Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция).
В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа.
При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1.
В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других.
Важно
Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза.
Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам.
Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов.
В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы.
Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные.
В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым.
Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии.
Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Совет
Проявления буллезного эпидермолиза разных типов объединяет одно – развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления.
При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается.
Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела.
Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции.
Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни.
У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы.
Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках.
Обратите внимание
После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей.
Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний.
Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза.
При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания.
На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы.
Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении.
Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза.
Важно
Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой.
Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода.
Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон.
Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей.
При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения.
Совет
Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии.
Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту.
Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи.
Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Буллезный эпидермолиз: причины, классификация, симптомы, основные методы диагностики и лечение
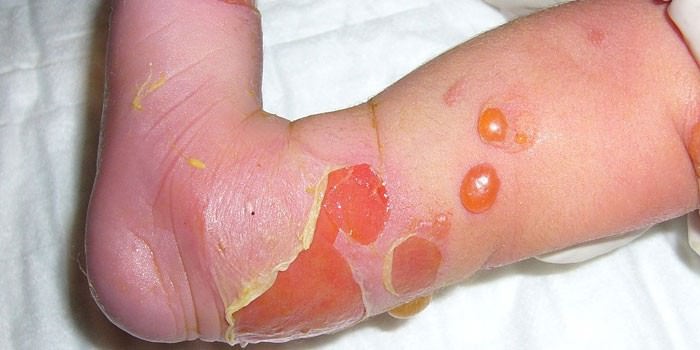
Иногда в литературе можно встретить диагноз «механобуллезная болезнь». Но в справочниках дерматологии ее нет.
На самом деле речь идет о буллезном эпидермолизе – гетерогенной группе генетических патологий, обусловленных мутациями определенных генов, которые ответственны за синтез структурных белков кожи.
Их основной признак — появление на коже и слизистых пузырей с серозным содержимым при малейшей механической травме. В последующем на месте буллезной сыпи образуются долго незаживающие эрозии.
Классификация
В зависимости от глубины поражения различают три основные формы заболевания и более 30 подтипов патологии, отличающихся по генотипу, фенотипу и характеру наследования.
В настоящее время эта классификация считается условной, поскольку на практике встречается очень много клинических форм, из-за чего часто возникают трудности с диагностикой. Специалисты стараются разработать более структурированную и приемлемую классификацию болезни, но пока безуспешно.
Причина
Заболевание развивается вследствие мутации в более чем десяти генах, в которых закодированы белки, составляющие структуру кожи. В большинстве случаев оно передается по наследству, реже является приобретенным, когда изменения в генах происходят спонтанно.
Ребенок может унаследовать патологию от одного из родителей. И даже если они не страдают буллезным эпидермолизом, взрослые могут быть скрытыми носителями мутировавших генов.
Условно все причины разделяют на две группы – генетические и внутриутробные. Основной патогенез – сбой в ферментной системе, в результате чего некоторые протеины становятся мишенью для атаки ферментов.
Генетические нарушения очень разнообразны:
- мутации в генах PLEC, KRT5 и KRT14 вызывают простую форму заболевания, атаке подвергаются протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1;
- мутации в генах LAMB3, LAMA3, LAMC2 провоцируют развитие пограничной формы патологии, мишенью становятся коллаген 17-го типа и ламинин-332;
- мутации в гене COL7A1 – причина дистрофической формы, при них происходит повреждение коллагена 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи;
- мишенью при синдроме Киндлера выступает белок киндлин-1.
Образование пузырей происходит в результате деструкции ключевых структурных белков и нарушения связи между клетками при малейшем механическом воздействии.
Внутриутробные причины, которые могут привести к мутации генов – самые банальные – курение, употребление алкоголя, прием лекарственных препаратов и воздействие других факторов, обладающих тератогенным эффектом.
Формы заболевания и его симптомы
Кожа пациентов не терпит никакого грубого механического или физического воздействия. Иначе в ответ быстро наступает отторжение эпидермиса и дермы. Сначала образуются пузыри, после вскрытия на их месте появляются эрозии. В некоторых случаях к высыпаниям может присоединяться вторичная инфекция.
Люди с диагнозом буллезный эпидермолиз находятся в группе риска по возникновению злокачественных новообразований.
Врожденный буллезный эпидермолиз может проявляться уже при рождении. В некоторых случаях кожа ребенка травмируется даже в момент прохождения по родовым путям матери.
Обратите внимание
Малышей, родившихся с такой патологией, называют «дети бабочки».
Однако первые симптомы могут появиться и значительно позже – точных сроков нет. Это может быть период новорожденности, младенчества или раннего детства. Реже всего заболевание начинается в юношестве, но такие случаи также зарегистрированы.
Приобретенный
Приобретенный буллезный эпидермолиз, как уже было сказано выше, возникает в результате спонтанных мутаций. Поэтому чаще всего первые признаки появляются уже у взрослых людей.
Пузыри и эрозии на коже пациентов, страдающих буллезным эпидермолизом, напоминают ожоги 3 степени.
Клиническая картина во многом схожа с симптомами этой болезни у детей и зависит исключительно от того, в каких генах произошли изменения.
Простой буллезный эпидермолиз
- ПБЭ подтипа Доулинга-Меара (герпетиформный). Наиболее тяжелая форма патологии. Первые признаки видны уже при рождении. Высыпания генерализованные, иногда образуют большие эрозии. Чаще всего поражается слизистая ротовой полости. Патология сопровождается герпетиформными высыпаниями на теле и конечностях. При этом подтипе могут страдать ногти. Происходит их отторжение. Впоследствии отрастают деформированные ногтевые пластинки или длинные ногти с гиперкератозом. Его проявления также возникают на ладонях и подошвах, позже они переходят в кератодермию.
Простой герпетиформный БЭ (Доулинг-Меара) - ПБЭ Кебнера (генерализованный подтип). Обширные высыпания появляются при рождении. Их излюбленная локализация – кисти и стопы. После заживления эрозий остаются участки депигментации. Иногда возникает ладонно-подошвенный гиперкератоз.
ПБЭ тяжелый генерализованный (Кебнера) - ПБЭ подтипа Вебера-Коккейна (локализованный). Наиболее легкая форма. Впервые заболевание проявляется в младенчестве, реже – в детстве или юношестве. Излюбленная локализация пузырей этого подтипа – ладони и стопы, реже – волосистая часть головы. Незначительные эрозии возникают в ротовой полости. Часто при патологии появляется гипергидроз ладоней и подошв. После заживления эрозий остается пигментация.
ПБЭ локализованный (Вебера-Коккейна) - ПБЭ Огна. Характеризуется сезонным образованием пузырей. Высыпания появляются летом, преимущественно на конечностях. Они сопровождаются онихогрифозом (искривлением ногтя в виде когтя) на больших пальцах ног.
- ПБЭ с мышечной дистрофией. Появление генерализованных пузырей при рождении. Сопровождается прогрессирующей мышечной дистрофией.
- ПБЭ с пятнистой пигментацией. Обширные участки с гиперпигментацией на туловище, кистях рук и стопах. При этом пузырей образуется немного, но по мере их регресса пигментация может усиливаться. Дополнительным симптомом этого подтипа являются веррукозные папулы – бородавчатые наросты на коже. Слизистая ротовой полости страдает незначительно.
- ПгБЭ Гертлица. Самая тяжелая форма буллезного эпидермолиза. Она приводит к летальному исходу в младенчестве или в раннем детском возрасте. Первые проявления – это обширные пузыри, которые возникают при рождении ребенка. Позже появляются участки гипертрофии грануляционной ткани, преимущественно в области рта, глаз, ноздрей, волосистой части головы и ушных раковин. Дополнительный симптом этого подтипа – утрата ногтевых пластинок и образование гипертрофической грануляционной ткани на концевых фалангах. Страдает ротовая полость – ямки в зубной эмали, обширные эрозии на слизистой ротоглотки. Возможно поражение эпителия носовой полости, слизистой конъюнктивы, пищевода, трахеи, гортани, прямой кишки и уретры. В тяжелых случаях развиваются многочисленные системные нарушения. Заболевание протекает на фоне анемии, происходит задержка роста и физического развития. К летальному исходу приводит осложнение патологии — сепсис.
Пограничный генерализованный тяжелый ВБЭ (летальный Херлитца) - ПгБЭ не-Гертлица. Высыпания в ротовой полости и поражение дыхательных путей не столь тяжелое, как при предыдущем подтипе. Однако встречаются тяжелые аномалии эпителиальной адгезии («склеивания»), что требует проведения трахеотомии с последующей установкой трахеостомы. Наиболее распространенные симптомы – участки сыпи на волосистой части головы и дистрофия ногтевых пластинок, а также периорифициальные (расположенные вокруг естественных отверстий организма, чаще всего – вокруг ануса) эрозии.
ПогрБЭ генерализованный среднетяжелый (не Херлитца) - Локализованный ПгБЭ (минимальный). Течение заболевания довольно легкое. Высыпания локализуются на кистях, стопах и передней поверхности голени. Реже развивается дистрофия ногтей, и появляются небольшие углубления на зубной эмали. На слизистой ротовой полости и носа образуются мелкие эрозии. Прогноз благоприятный.
- ПгБЭ с атрезией (отсутствием или заращением) привратника. Еще одна тяжелая форма заболевания. Пациенты страдают от неимоверной хрупкости кожных покровов и слизистых. У них часто обнаруживают аномалии развития органов мочевыделения, например, гидронефроз или нефрит. Отличительная особенность – рудиментарные ушные раковины.
- Локализованный доминантный ДБЭ (ДДБЭ, подтип Коккейна-Турена). Пузыри образуются на участках, которые чаще всего подвержены трению – колени, область крестца и акральные поверхности. Впоследствии на месте сыпи кожа подвергается дистрофическим процессам, рубцеванию с образованием милиумов. Страдают ногти, вплоть до полного отторжения и атрофического рубцевания последних фаланг пальцев.
- Генерализованный доминантный ДБЭ (ДДБЭ, подтип Пазини). Течение патологии более тяжелое, чем у локализованной формы. Пузыри сменяются эрозиями. После их заживления образуются рубцовые бляшки и милиумы. Дополнительный симптом – спонтанное появление на теле бесцветных плотных папул. По мере взросления пациента генерализованные очаги регрессируют в локализованные с преимущественным поражением кожи конечностей. Заболевание сопровождается дистрофическим изменением ногтей, часто приводящим к утрате ногтевой пластинки, и образованием небольших эрозий на слизистой ротовой полости.
- Рецессивный ДБЭ (РДБЭ). Локализованная форма отличается средней степенью тяжести и по симптомам схожа с ДДБЭ Коккейна-Турена. Подтип Аллопо-Сименса – РДБЭ с тяжелым течением. При рождении возникают обширные генерализованные высыпания. Иногда у ребенка развивается масштабное отслоение кожи (синдром Барта). Чередование обострений и периодов нестойкой ремиссии приводит к распространенному рубцеванию, в результате чего часто образуются сгибательные контрактуры. Заболевание сопровождается проявлением псевдосиндактилии – пальцы ребенка находятся в своеобразной «варежке», что может привести к их сращению. Периорифицивального поражения нет, очаги высыпаний локализуются на голове и шее. Особенно страдает волосистая часть головы, где быстро прогрессирует алопеция. Одно из самых серьезных проявлений – обширное поражение слизистой ротоглотки. После эрозий образуются рубцы, которые ограничивают движения языка и мешают нормально открывать рот. На зубной эмали появляются точечные уплотнения, часто приводящие к кариесу и даже полной потере зубов. Очаги патологии на слизистой верхних дыхательных путей провоцируют стеноз, из-за чего пациенту часто приходится ставить трахеостому. Эрозии в пищеводе вызывают образование стриктур. Сочетание всех этих проявлений приводит к недоеданию и как следствие – задержке роста. У таких детей часто развивается анемия.
Рецессивный дистрофический тяжелый генерализованный БЭ (Аллопо-Сименса)
Наиболее часто у пациентов с РДБЭ, которые достигли полового созревания, развивается тяжелое осложнение – плоскоклеточная карцинома. Эта патология отличается агрессивным течением, склонностью к инвазии и быстрому метастазированию.
Первые признаки видны уже при рождении. Поскольку высыпания одновременно затрагивают различные ультрастуктурные уровни, клинические проявления могут напоминать как дистрофическую, так и пограничную форму буллезного эпидермолиза. В процессе заживления эрозий на коже происходят атрофические изменения.
По мере взросления буллезные высыпания проходят, но развивается пойкилодермия. Это проявления атрофии, гиперкератоза, сосудистых и пигментных нарушений на коже открытых участков тела, то есть тех, которые подвержены инсоляции. И чаще всего они возникают у пациентов с фоточувствительностью. Помимо этого патология затрагивает ногти и может привести к синдактилии пальцев рук и ног.
Со временем течение заболевания осложняется нарушениями со стороны внутренних органов. Возникают воспалительные процессы на слизистой ротовой полости и стриктуры в пищеводе и уретре.
Диагностика
Анамнез и клиническая картина позволяют установить только предварительный диагноз. Окончательно подтвердить буллезный эпидермолиз могут только методы лабораторной диагностики. Для этого проводят биопсию и образцы кожи подвергают ряду специфических тестов.
- Трансмиссионная электронная микроскопия. Помогает увидеть и оценить определенные элементы кожи, а также обнаружить в ней ультраструктурные изменения.
- Иммунофлюорисцентное антигенное картирование (ИАК). Определяет уровень образования пузыря. Позволяет оценить количество структурных белков и определить, какой из них стал мишенью, а также уточнить ферментную активность ткани. Уменьшение уровня ключевого протеина может свидетельствовать о его низком синтезе или массовом разрушении.
- Генетический анализ (молекулярная или ДНК-диагностика). Завершающий анализ. Прямое секвенирование (определение последовательности соединения нуклеотидов или аминокислот в цепи ДНК или РНК) дает возможность точно определить форму буллезного эпидермолиза и его тип наследования.
Если в семье есть родственники, страдающие буллезным эпидермолизом, целесообразно пройти генетическую консультацию на стадии планирования беременности, а также пренатальную диагностику, которая позволит выявить патологию на раннем этапе развития плода.
Методы лечения
Буллезный эпидермолиз – системная патология, поэтому лечением занимается мультидисциплинарная команда врачей.
Заболевание полностью излечить невозможно, поскольку специфические препараты для борьбы с ним до сих пор не найдены.
Однако современные методы симптоматической терапии, правильный уход за ранами, а также превентивные меры значительно облегчают состояние пациентов и дают им возможность прожить довольно долго.
Цель всех лечебных мероприятий:
- предупреждение разрастания и вскрытия пузырей;
- быстрое заживление эрозий и эпителизация кожи;
- предупреждение развития осложнений.
В качестве симптоматической терапии могут быть назначены следующие группы препаратов:
- анальгетики устранят боль;
- антигистаминные избавят от зуда и жжения;
- антибиотики помогут при вторичном инфицировании эрозий;
- глюкокортикоиды снимут воспаление и неприятные ощущения на коже;
- поливитамины окажут общеукрепляющее действие.
Наружное лечение и уход за кожей заключается в обработке пузырей антисептиками, наложении повязок, в основе которых неприлипающие материалы и несколько слоев бинтов. Кроме того, проводят УФО – профилактическое облучение кожи ультрафиолетовыми лучами для предупреждения инфицирования эрозий.
Продолжительность жизни с буллезным эпидермолизом
Прогноз и продолжительность жизни больного буллезным эпидермолизом зависит от многих факторов — формы патологии и ее подтипа, качества ухода, наличия осложнений и общего состояния пациента.
Раньше летальный исход наступал в младенчестве, реже малыши доживали до трех лет.
Простой буллезный эпидермолиз и некоторые локализованные формы других типов имеют наиболее благоприятный прогноз. Иногда проявления болезни регрессируют, и пациенты проживают обычную жизнь. Добиваясь стойкой ремиссии, они никогда не забывают о мерах профилактики и соблюдают все рекомендации врачей.
Наиболее неблагоприятный прогноз при дистрофической форме заболевания. Множественные осложнения часто приводят к летальному исходу еще в детстве.
Важно
В настоящее время, к сожалению, не все родители могут обеспечить ребенку-бабочке надлежащий уход. Однако специальные благотворительные фонды не оставляют малышей и помогают им по мере возможности.
Буллезный эпидермолиз: симптомы у взрослых и детей, причины, лечение, препараты, отзывы

Генетическое заболевание, которое проявляется в виде отсутствия защищенности кожи от механических соприкосновений и потираний — кожа реагирует высыпаниями, — называют буллезный эпидермолиз. Проблема имеет наследственный характер и не зависит от инфекционных заболеваний, перенесенных во время вынашивания ребенка.
Особенности патологии
Заболевание проявляет себя, когда случается мутация генов в количестве более десяти. Любое механическое воздействие, при наличии заболевания, приводит к повреждениям кожи. Обычно наблюдается появление пузырей.
У детей определяется патология в раннем возрасте. Как проявляет себя заболевание зависит от его вида по классификации и еще подвида. При типе болезни синдром Киндлера новорожденные появляются на свет с симптомами буллезного эпидермолиза с уже сформированными пузырями.
Заболевание встречается в одинаковом соотношении у представителей женского пола и мужского. В детском возрасте обращают внимание на возможное присутствие такой особенности: врожденную неполноценность или отсутствие эластических волокон, что будет указывать на заболевание.
Этот видеоролик содержит полезную информацию об особенностях буллезного эпидермолиза:
Классификация
Составить классификацию и разделить буллезный эпидермолиз (сокращенно БЭ) на определенный виды удалось с применением электронно-микроскопического исследования. Он помог увидеть разные уровни поражения кожи, и специалисты взяли эту информацию в основу классификации.
Различают три типа заболевания и четвертый вид, который рассматривают отдельно:
- простой вид БЭ – так обозначены случаи, когда локализация пузырей находится на поверхности эпидермиса;
- пограничный вид БЭ – пузыри располагаются на уровне, который называют светлой пластинкой;
- дистрофический вид БЭ – такое название получает заболевание в том случае, когда пузыри расположены в сосочковом слое, в верхней его части.
- разновидность БЭ — синдром Киндлера – нет закономерностей в локализации пузырей, их образование наблюдается на разных уровнях.
Каждый вид болезни специалисты подразделяют на подвиды. Они различаются по тому признаку, какие мутировали гены и типом наследования.
Причины возникновения
Буллезный эпидермолиз передается по наследству. Один из родителей, может быть такое, что и оба родителя имеют мутацию определенных генов. При этом они могут быть вполне здоровыми людьми.
У зачатого ребенка, при определенных сочетаниях генетики родителей, происходит унаследование болезни. Тип проблемы зависит от пути наследования: рецессивного или доминантного. Речь идет о форме взаимодействии пары генов. Эти нюансы, которыми хорошо владеют генетики, будут влиять на тип и подтип заболевания.
Симптомы
Основным признаком болезни является склонность организма к возникновению эрозий после даже самого незначительного воздействия на кожу и слизистые поверхности. Характерно появление пузырей. Высыпания могут произойти и без видимой причины.
Пузыри имеют такие особенности:
- полости наполнены прозрачным содержимым,
- иногда пузыри имеют наполнение геморрагической жидкостью,
- пузыри выглядят напряженными.
Локализация пузырей может быть различной:
- стопы и кисти рук,
- ротовая полость,
- ногтевые валики,
- подмышечная полость,
- вся поверхность кожи.
В зависимости к какому виду и подвиду поражения относится болезнь, пузыри могут заживать:
- с появлением рубцов,
- не оставляя следа.
Диагностика
Для определения вида заболевания применяются три метода:
Врожденный буллезный эпидермолиз у детей (фото)
Лечение буллезного эпидермолиза
Помощь пациентам, страдающим БЭ, предусматривается комплексная. Патология относится к системному заболеванию, поэтому лечение оказывается согласованными действиями команды врачей.
Терапевтическим способом
Симптоматическое лечение заключается в поддержании состояния кожи с целью:
- не допустить увеличения площади пузырей,
- создать условия, при которых не случится нарушение целостности пузыря, потому что такая ситуация приводит к эрозии поверхности кожи;
- если имеются раны, то за ними обеспечивается квалифицированный уход, часто используются специальные лечебные повязки, которые не пристают к ране. При наличии ран основная задача сводится к их заживлению и созданию условий для эпителизации.
В этом видео будет рассказано о человеке с рассматриваемой патологией:
Медикаментозным способом
Применяются препараты такого направления:
- анальгетики,
- витамины,
- антибиотики,
- препараты, работающие на генном уровне и регулирующие происходящие процессы.
Другие методы
Современные способы лечения:
- Использование стволовых клеток, чтобы повлиять оздоравливающе на вновь появляющиеся клетки.
- Протеиновая терапия – пополнение недостающего в организме белка, чтобы восстанавливать коллагеновые волокна.
- Комбинированная терапия, когда используют кожные трансплантаты.
- Клеточная терапия – пациенту вводят клетки донора со здоровыми генами, которые кодируют белок.
- Генная терапия – пациенту вводится ген, который у него поврежден, для кодирования белка.
Профилактика заболевания
Меры по предупреждению обострений болезни:
- Для больного подбирают одежду, обувь, предметы обихода, которые исключают травмирование при контакте.
- Пациенту рекомендуют соблюдать специальную диету, содержащую достаточное количество легкоусвояемого белка.
- Наблюдение врача и профилактические курсы лечения.
Осложнения
В случае образования ран возможны осложнения из-за их инфицирования.
Некоторые формы заболевания ведут к поражению внутренних органов. Бывают такие нарушения:
- сужение пищевода,
- запоры,
- поносы,
- непроходимость пищевода,
- болезни прямой кишки,
- нарушения глотания,
- заболевание желудка,
- повреждение роговицы,
- дистрофия мышц,
- выворот век,
- болезни зубов,
- кардиомиопатия,
- сращение пальцев,
- поражение ногтей.
Прогноз
Генетическое заболевание не получается полностью вылечить. Необходимо быть под наблюдением врачей, чтобы не допускать ухудшения ситуации. Большое значение имеет тот факт, какая разновидность болезни у пациента.
Тяжелые формы болезни способны привести к таким результатам:
- инвалидность еще в раннем возрасте,
- летальный исход новорожденных детей в течение нескольких месяцев или даже дней.
Формы болезни, которые относят к легким, дают возможность пациенту полноценно жить, но при условии регулярного поддерживающего лечения и соблюдения необходимой профилактики.
В этом видео собраны советы педиатров в отношении буллезного эпидермолиза:
Буллезный эпидермолиз: 4 основных типа, характерные симптомы и 7 основных принципов лечения

Врождённый буллёзный эпидермолиз — генетическое заболевание, которое характеризуется многообразием форм, но объединяет их поражение кожного покрова.
Частота выявления буллёзного эпидермолиза составляет 1:300000 новорождённых.
Причины возникновения заболевания
Буллёзный эпидермолиз — врождённое, наследственное, генетическое заболевание. Развивается оно вследствие мутации генов, которые кодируют специальные белки кожи, обеспечивающие связь между дермой и эпидермисом. Такую функцию выполняют около 15 генов, способных к новым мутациям.
Провоцирующими факторами для развития заболевания могут быть:
- травмы кожного покрова;
- изменения температуры окружающей среды;
- неудобная одежда или обувь.
Трудностью для диагностики является то, что каждая форма заболевания имеет различные причины. Простой буллёзный эпидермолиз вызывается мутацией KRT5 и KRT14 генов.
Такой вид заболевания наследуется по аутосомно-доминантному механизму наследования. При данной ситуации происходит нарушение баланса между белками и ферментами.
При повреждении кожного покрова структурные белки разрушаются, и развивается отторжение эпидермиса.
Более тяжёлой формой заболевания является пограничная. Такой вид буллёзного эпидермолиза вызывается мутацией в генах LAMB3 и LAMA3. Данная форма наследуется по аутосомно-рецессивному типу. Страдают белки коллагена. Поэтому, помимо образования пузырей, кожный покров при такой форме склонен в повышенной ломкости.
Дистрофический тип буллёзного эпидермолиза может наследоваться как по аутосомно-доминантному типу, так и по аутосомно-рецессивному. Мишенью поражения становится коллаген 7-го типа, кодирующийся дефектным геном COL7A1.
Такой коллаген отвечает за состояние соединительной ткани во всех органах и системах.
Поэтому вместе с поражением кожного покрова у пациентов выявляются контрактуры суставов, злокачественные новообразования, которые могут развиваться на месте рубцов (заживших эрозий).
Механизм развития заболевания
Механизм развития некоторых форм заболевания имеет незначительные особенности, но в целом патогенез буллёзного эпидермолиза сводится к нарушению функционирования ферментов под воздействием генных мутаций.
Такие изменения разрушают белки, необходимые для постройки прочных связей между слоями кожи.
Если связи непрочны или вовсе отсутствуют, то клинически это проявляется отторжением верхних слоёв эпидермиса, иными словами, образованием пузырей.
Классификация
На сегодняшний день выделено 4 основных типа и 6 подтипов заболевания. Внутри подтипов существует около 30 клинических форм.
Клиническая картина
Для каждого типа заболевания характерно появление пузырей.
Для всех форм буллёзного эпидермолиза характерно наличие пузырей. Они имеют шаровидную форму, всегда наполнены жидкостью.
Пузыри образуются в местах трения или травмы, для повреждения кожного покрова хватает незначительного воздействия извне. Для дистрофической формы характерно появление вялых пузырей, жидкость смещается в зависимости от положения тела.
Клинических форм существует большое количество, но важно иметь представление о наиболее часто встречающихся типах заболевания.
Совет
Среди простого буллёзного эпидермолиза следует выделить: тяжёлый генерализованный (синдром Кебнера), локализованный (синдром Вебера — Коккейна), герпетиформный (синдром Доулинг — Меара).
Синдром Кебнера. Заболевание выявляется в раннем возрасте практически с первых дней жизни. На коже ребёнка появляются генерализованные обширные пузыри. Чаще начинаются со стоп.
Далее с возрастом наблюдаются утолщённые участки на стопах, повышенная влажность ладошек. Ухудшается состояние кожного покрова летом, так как тёплое время года негативно влияет на кожу, вызывая повышенную потливость. В зимнее время отмечается значительное улучшение.
В период полового созревания и климакса у женщин состояние кожи также заметно улучшается.
Синдром Вебера — Коккейна появляется чаше в возрасте, когда ребёнок начинает вставать на ножки или ходить. Называется такой синдром локализованным, так как поражение кожного покрова приходится на стопы и ладони.
Данная форма заболевания характеризуется образованием пузырей в ответ на травматизацию кожного покрова. В более сознательном возрасте, когда малыш играет игрушками или хлопает в ладоши, появляются пузыри на местах трения или травм. Прогрессирование заболевания наблюдается до 4 — 5-летнего возраста.
Затем в период полого созревания отмечается улучшение. Потливость ладошек и стоп сохраняется длительное время.
Синдром Доулинг — Меара. Множество пузырей на коже можно уже наблюдать в первые дни жизни. Характерным отличием служит появление изменений на слизистой оболочке полости рта, а также отсутствие связи с травматизацией. Пузыри склонны к группировке.
Данная форма заболевания быстро развивается до 1 года жизни ребёнка, затем можно увидеть положительную динамику, даже полное исчезновение симптомов болезни. Нередко можно визуализировать гнойное содержание пузырей и формирование корочек.
После их исчезновения остаётся стойкая пигментация. Спустя 2 — 3 года болезни можно чётко отметить связь со временем года (летом ухудшение, зимой улучшение). Помимо кожных проявлений часто можно наблюдать симптомы: бледность, вялость, головокружения, запоры, анемию.
Такое заболевание является фактором риска для развития базальноклеточного рака кожи.
Среди пограничного типа буллёзного эпидермолиза наиболее популярными являются клинические формы: генерализованная тяжёлая летальная (синдром Герлитца), среднетяжёлая, с поздней манифестацией.
Пограничный генерализованный тяжёлый летальный синдром Герлитца. Данный тип заболевания характеризуется полиморфизмом клинических проявлений. Начинается появление симптомов с первых дней жизни или сразу после рождения.
На кожном покрове и слизистой оболочке полости рта новорождённого ребёнка наблюдаются множественные, вялые, быстро вскрывающиеся пузыри. Придатки кожи также подвергаются изменениям. Отмечается деформация ногтей или их отсутствие. При прорезывании зубов выявляется гипоплазия эмали.
Заживление пузырей на слизистой полости рта не проходит бесследно, оставляя сужение ротовой щели. Поражение глаз может приводить к сращению век. В более старшем возрасте развивается диффузная алопеция (поредение волос), плоскоклеточный рак кожи. При таком заболевании поражается довольно большое количество органов и систем.
Обратите внимание
Это приводит к отставанию в физическом развитии, анемии, нарушению дыхания, пневмонии, сепсису. Именно последние проявления заболевания приводят к летальному исходу. Смертность при данной форме крайне высока.
Пограничный генерализованный среднетяжёлый буллёзный эпидермолиз. Начальные проявления не отличаются от тяжёлой формы заболевания. Также начинаются с появления пузырей на кистях и стопах.
Но разрешение патологического процесса происходит быстрее, кожный покров эпителизируется, и состояние кожи заметно улучшается со временем.
Некожные симптомы также встречаются, но риск смертельного исхода значительно ниже.
Пограничный буллёзный эпидермолиз с поздней манифестацией.
Отличительной особенностью является то, что данная форма проявляется не в период новорождённости, а в дошкольном или школьном возрасте.
Также появление пузырей начинается с ладоней или стоп, реже на других участках кожи. Ногти могут отсутствовать. Выявляют гипоплазию зубной эмали. Атрофия кожного покрова развивается медленно.
Дистрофический тип буллёзного эпидермолиза делится на 2 варианта: доминантный и рецессивный, при этом последний имеет несколько клинических форм.
Доминантный дистофический буллёзный эпидермолиз. Как правило, дебютирует заболевание после первого месяца жизни. Могут появляться пузыри спонтанно, но чаще они образуются в местах трения. К 5 годам новые элементы практически не наблюдаются.
Проявляются лишь после значительной травмы или ушиба или на месте заживления давних эрозий. Полное улучшение кожного покрова регистрируется в возрасте 10 лет. Период полового созревания также способствует окончательному восстановлению кожи.
Ногтевые пластинки, которые были поражены в период активного появления пузырей, вновь не восстанавливаются.
Рецессивный дистрофический тяжёлый генерализованный буллёзный эпидермолиз (синдром Аллопо-Сименса). Данный синдром среди дистрофических форм буллёзного эпидермолиза является самым тяжёлым. Заболевание регистрируется с самого рождения. Нередко дети появляются на свет без эпидермиса.
Важно
На коже новорождённого появляется множество пузырей и эрозий, которые могут заживать в течение нескольких месяцев. На месте заживших поражённых участков появляются рубцы, ограничивающие движения. Слизистая оболочка также подвержена образованию пузырей, эрозий и рубцеванию.
Перерождение слизистой во рту ведёт к атрофии сосочков языка, сужению ротовой щели, дефекту речи. Рубцевание в пищеводе ведёт к его сужению и, следствием этого является нарушение глотания, пищеводный рефлюкс (обратный запрос пищи).
При изменениях слизистой оболочки прямой кишки происходит сужение просвета органа с явлениями запора.
Помимо вышеперечисленных проявлений, у больного можно отметить выпадение зубов, почечную недостаточность, остеопороз, деформации кистей и стоп, боли в суставах и мышцах, анемию, отставание в физическом и нервно-психическом развитии. Такие проявления болезни существенно ухудшают качество жизни ребёнка. Отсроченным проявлением может быть плоскоклеточный рак кожи.
Рецессивный дистрофический инверсный буллёзный эпидермолиз. Проявляется заболевание с первых недель жизни ребёнка. По мере взросления пациента развивается алопеция, приводящая к полному исчезновению волос на теле. Поражение слизистых оболочек ведёт к сужению поражённых органов. К 30 годам у больного развивается плоскоклеточный рак кожи.
Синдром Киндлера. Клинические проявления заболевания не отличаются от типичных симптомов буллёзного эпидермолиза, но выраженность их превосходит все формы.
Заболевание характеризуется поражением всех слоёв кожи и слизистой оболочки. Поражаются все органы и системы. Деформируются суставы, конечности, пальцы.
Например, после заживления эрозий, кожа фаланг пальцев рубцуется так, что фиброзные перетяжки способствуют к самопроизвольной ампутации.
Осложнения буллёзного эпидермолиза встречаются довольно часто. Наиболее серьёзным последствием является плоскоклеточный рак кожи. Он развивается на наиболее поражённых участках кожного покрова и склонен к быстрому метастазированию.
Диагностика заболевания
На сегодняшний день пренатальная диагностика (во время беременности) буллёзного эпидермолиза существует и заключается она в проведении генетического анализа.
Гистологическое исследование поражённого участка кожи позволяет убедиться в наличии заболевания, но не определяет форму буллёзного эпидермолиза.
Для более точной диагностики требуется углублённое исследование материала методом непрямой иммунофлюоресценции. Данная процедура требует соблюдения ряда правил сбора биоптата. Материал забирают на границе видимо здоровой кожи со свежим пузырём. Такой метод позволяет определить уровень поражения и, следовательно, установить форму заболевания.
Пациенту требуется постоянный контроль общего анализа мочи и крови, а также регулярная консультация офтальмолога, гастроэнтеролог, кардиолога, хирурга, гинеколога, уролога, стоматолога, оториноларинголога, онколога при наличии осложнений.
Принципы лечения
Специфического лечения буллёзного эпидермолиза не существует!
Основные принципы направлены на уход за поражённой поверхностью кожного покрова и слизистых оболочек, лечение осложнений.
Показания к госпитализации:
- распространённое поражение кожи и слизистых оболочек;
- наличие осложнений заболевания, в зависимости от их характера, пациент может быть госпитализирован либо в хирургическое отделение, либо соматическое.
Пузыри вскрывают иглой, обработанной раствором антисептиков. Прокол рекомендуется в 2 местах для выхода жидкости, при этом, не задевая дно пузыря. Содержимое удаляют марлевыми салфетками. Важно не делать резких движений и аккуратно, промакивающими движениями, убирать жидкость.
Затем необходима обработка раствором антисептика (водные растворы: хлоргексидина биглюконат 0,05%, метилтиониния хлорид 1%, гидроксиметилхиноксалиндиоксид 1%). После обработки пузырей накладывается специальная стерильная повязка.
Перевязочный материал должен быть без клеевой основы, чтобы не происходило прилипание к коже больного. Предпочтение отдают липидо-коллоидным повязкам с восковым покрытием, а также с гидрогелевым или силиконовым покрытием.
Совет
Если происходит инфицирование поражённых участков кожи, то следует использовать перевязочный материал с антимикробным и антисептическим действием. Для фиксации используют вторичные перевязки в виде трубчатых бинтов.
В случае прилипания повязки к коже ребёнка, рекомендуется наносить очиститель для кожи в виде спрея, для профилактики болезненных ощущений у пациента. Для быстрой эпителизации и регенерации эрозий используют декспантенол. Обработка слизистой оболочки включает в себя полоскание раствором хлоргексидина и использование геля Холисал.
Профилактика
Самым лучшим профилактическим мероприятием для всех заболеваний является пренатальная диагностика. Медико-генетическое консультирование поможет избежать тяжёлых заболеваний и оценить риск рождения больного ребёнка.
При развитии заболевания следует избегать травматизации кожи, повышенного потоотделения. Носить свободную одежду и обувь из качественных и натуральных материалов. Уход за кожей, своевременная смена повязок уменьшает площадь поражения. Для своевременного выявления осложнений требуется постоянная консультация специалистов и регулярная сдача анализов.
Заключение
Несмотря на то, что буллёзный эпидермолиз — заболевание с серьёзным прогнозом, на сегодняшний день существует пренатальная диагностика, которая позволяет определить наличие тяжёлой патологии. Проблема данной болезни заключается в постоянной потребности перевязочных материалов, которые имеют высокую стоимость.
Но только специальные повязки способны улучшить качество жизни больного, предотвратить контрактуры, и, следовательно, инвалидизацию пациента.
Мы приложили много усилий, чтобы Вы смогли прочитать эту статью, и будем рады Вашему отзыву в виде оценки. Автору будет приятно видеть, что Вам был интересен этот материал.
Спасибо!
(2
Comments
(0 Comments)