Болезнь шарко мари тута: причины, 8 симптомов, 3 метода лечения
Болезнь Шарко Мари Тута: причины, 8 симптомов, 3 метода лечения
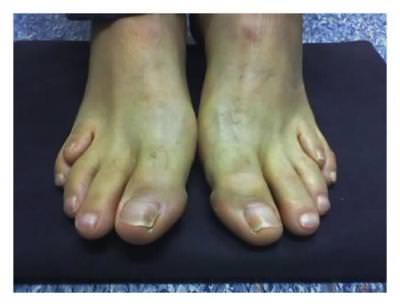
Шарко Мари Тута болезнь относится к разряду наследственных невропатий, для которых характерно поражение волокон периферической нервной системы, причём как сенсорных, так и моторных. Нередко можно встретить и другие названия патологии. К примеру, перонеальная или невральная амиотрофия Шарко-Мари.
Данная нозология считается одним из наиболее частых представителей наследственных заболеваний, так как встречается у 25-30 человек на 100 тыс. населения.
В современной неврологии выделяют 4 варианта недуга. Но скорее всего это не окончательное число. Ведь до сих пор не известны все виды мутаций в генах, которые способны привести к заболеванию.
Синдром Шарко первого (или демиелинизирующего) типа наблюдается у 60% пациентов. Он дебютирует до двадцатилетнего возраста, чаще даже в первые годы жизни ребёнка. Передаётся заболевание аутосомно-доминантно.
Второй вариант патологии, именуемый также аксональным и встречающийся в 20% случаев, наследуется таким же образом. То есть когда один из родителей болен, дети в любом случае «получат» данную генетическую «поломку» и она разовьётся затем в заболевание.
Но у последнего вида амиотрофии манифестация симптомов порой откладывается до 60-70 лет.
Обратите внимание
Эти два типа болезни Шарко Мари Тута отличаются тем, что при первом происходит разрушение миелиновой оболочки нерва, а при втором – в первую очередь повреждается аксон.
Третьим вариантом патологии является тот, который наследуется через Х хромосому доминантно. При этом у мужчин симптомы выражены в большей степени, чем у женщин. Задуматься об этом варианте синдрома врача заставит информация о том, что признаки недуга обнаруживаются у мужчин не в каждом поколении семьи.
Невральная амиотрофия четвёртого типа считается демиелинизирующей и наследуется аутосомно-рецессивно, то есть существует шанс рождения здоровых детей у больных родителей.
Основной и единственной причиной любого типа болезни Шарко-Мари-Тута являются точечные мутации в генах.
На данный момент науке известны около двух десятков генов, которые подвергаются изменениям, приводящим впоследствии к амиотрофии.
Чаще всего «поломки» обнаруживаются в генах, расположенных на 1-й, 8-й хромосомах, а именно PMP22, MPZ, MFN2 и др. И это лишь часть из них.
Многие остаются так сказать «за кадром», ведь около 10-15% пациентов даже не догадываются о своей болезни и не обращаются за медицинской помощью.
Демиелинизирующий тип патологии также связывают с приобретенной аутоиммунной агрессией, при которой клетки, создающие наружную оболочку нерва, воспринимаются организмом как чужеродные и подвергаются уничтожению.
Существуют также факторы риска, способствующие более яркому проявлению симптомов болезни Шарко-Мари-Тута и усугублению уже имеющейся клинической картины.
К триггерам недуга относят:
- употребление алкоголя;
- применение потенциально нейротоксичных препаратов. К примеру, винкристина, солей лития, метронидазола, нитрофуранов и т.п.
Больным, страдающим наследственной перонеальной амиотрофией противопоказан наркоз, при котором используют тиопентал.
Болезнь Шарко-Мари-Тута проявляется даже в пределах одной семьи не всегда одинаково. И дело не в многообразии её признаков. А в том, что гены, кодирующие данную патологию, способны с разной степенью выраженности формировать симптомы. Проще говоря, имея идентичную «поломку» в хромосомах, признаки недуга у отца и сына будут иметь индивидуальную окраску.
Общие симптомы
Клиническая картина заболевания практически не зависит от его типа и включает:
- атрофию мышц дистальных, то есть наиболее отдалённых от туловища, отделов конечностей;
- снижение сухожильных и периостальных рефлексов;
- изменение чувствительности, характеризующееся её выпадением, но никогда не сопровождающееся появлением ощущения покалывания или «ползания мурашек»;
- деформацию опорно-двигательного аппарата – сколиоз, увеличение свода стопы и др.
Но всё же существует ряд признаков несколько отличающих течение заболевания при различных его вариантах.
Первый тип
Болезнь Шарко-Мари-Тута первого типа нередко протекает в исключительно стёртой форме, при которой пациенты не ощущают изменений в организме и не обращаются за медицинской помощью вообще. Если же патология себя проявила, то происходит это в период первого, максимум второго, десятилетия жизни.
При этом наблюдаются:
- болезненные судороги в мышечном массиве голени, причём редко в икроножной мышце, чаще в передней группе мышц. Подобные спазмы нарастают после периода длительной физической нагрузки (ходьбы, занятий спортом, езде на велосипеде);
- изменения в походке, связанные с постепенным усилением слабости в мышцах. При этом у детей это может дебютировать в хождении на цыпочках;
- деформация стоп с формированием высокого свода последних и наличием молоточкообразных пальцев, которая развивается как результат дисбаланса в тонусе сгибателей и разгибателей;
- атрофия мышц, начинающаяся со стоп и поднимающаяся на голень. Затем процесс затрагивает кисть – появляется тремор в руках и выраженная слабость в пальцах, особенно при попытке выполнять мелкие движения. К примеру, застёгивать пуговицы, перебирать крупу;
- угнетение или полное отсутствие сухожильных и периостальных рефлексов, а именно ахиллового, карпорадиального, при сохранных с более проксимальных отделов рук и ног. То есть коленный и рефлексы с двуглавой и трёхглавой мышцы остаются интактными;
- нарушения чувствительности в кистях и стопах, выражающиеся в её постепенном выпадении. Причём стартует патология с вибрационной и тактильной сферы, распространяясь на суставно-мышечные и болевые ощущения;
- сколиоз и кифосколиоз;
- утолщение нервных стволов, чаще всего поверхностного малоберцового и большого ушного.
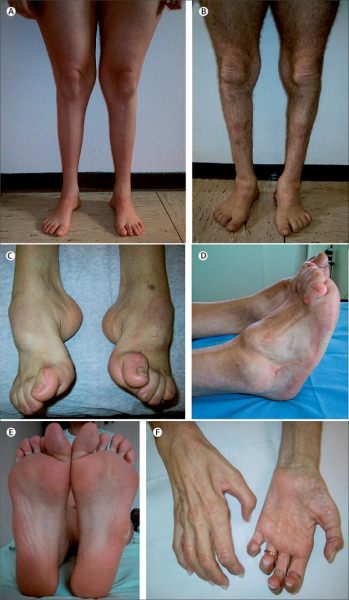
Для болезни Шарко характерна атрофия мышц именно в дистальных отделах конечностей. При этом если не выражена подкожно-жировая клетчатка, объём голени и бедра разительно отличается, и ноги приобретают вид таковых у аиста или походят на перевёрнутую бутылку для шампанского.
Невральная амиотрофия Шарко Мари первого типа имеет атипичные формы.
Одна из них — синдром Руси-Леви, при котором наблюдается выраженный тремор при попытке удержать руки в одном положении и неустойчивость при ходьбе.
Сюда же относится заболевание, проявляющееся, помимо стандартных симптомов, парезами, гипертрофией мышц голени, резким выпадением чувствительности и ночными судорогами в икроножных мышцах.
Второй тип
Для болезни Шарко-Мари-Тута второго типа, кроме более позднего начала, характерны:
- менее выраженные изменения чувствительности;
- более редкая встречаемость деформаций стопы и пальцев;
- наличие синдрома беспокойных ног (возникают неприятные ощущения в ногах во время отхода ко сну, заставляющие пациента двигаться, что облегчает состояние);
- нередко сохранная сила в кисти;
- отсутствие утолщения нервных стволов.
При синдроме Шарко-Мари-Тута, передающемся через Х-хромосому, могут встречаться нейросенсорная тугоухость (снижение слуха) и транзиторная энцефалопатия, возникающая после физической нагрузки на высоте.
Для последней характерно появление симптомов через 2-3 дня после занятий. Признаками патологии становятся шаткость, нарушение речи, глотания, слабость в проксимальных отделах рук и ног.
Обычно клиническая картина недуга исчезает самостоятельно в течение пары недель.
Что нужно для подтверждения диагноза
Необходимо помнить, что в первую очередь заподозрить именно болезнь Шарко-Мари-Тута врачу позволит отягощённый семейный анамнез, то есть наличие сходных симптомов у кого-то из ближайших родственников пациента.
Из инструментальных методов обследования наиболее информативным является электронейромиография (ЭНМГ). При этом обнаруживается снижение скорости проведения импульса по нервам с нормальных 38 м/с до 20 м/с на руках и 16 м/с на ногах. Исследуют также сенсорные вызванные потенциалы, которые не вызываются вовсе или имеют значительно уменьшенную амплитуду.
При втором типе синдрома Шарко изменения на ЭНМГ не возникают. Наблюдается патология лишь при исследовании вызванных сенсорных потенциалов.
Важно
Окончательно удостовериться в наличии невральной амиотрофии позволяет биопсия нерва с гистологическим анализом полученных материалов.
При этом для первого варианта болезни характерны:
- демиелинизация и ремиелинизация волокон с формированием «луковичных головок»;
- снижение доли крупных миелинизированных волокон;
- атрофия аксонов с уменьшением их диаметра.
А второй тип болезни Шарко Мари Тута не сопровождается демиелинизацией и образованием «луковичных головок», в остальном признаки совпадают.
Подходы к лечению болезни Шарко
На данный момент не существует специфической терапии при болезни Шарко, разработана лишь симптоматическая. Причём в большей степени упор делается на ЛФК и физиолечение. Не менее важным этапом считается правильная гигиена стоп и ношение ортопедической обуви.
Хирургическое лечение
Из хирургических вмешательств при синдроме Шарко чаще всего прибегают к артродезу голеностопных суставов: удалению хрящевых поверхностей и сращиванию таранной и большеберцовой костей. Это сопровождается фиксированием ноги в одном положении и утратой подвижности стопы, но позволяет сохранить возможность передвигаться.
Физиотерапевтические методы
При выявлении признаков патологии сразу же прибегают к обучению пациента упражнениям ЛФК, которые позволят как можно дольше сохранять эластичность мышц и сухожилий. Также благотворно влияют на состояние больных массаж и аэробные нагрузки, к примеру, плавание, спортивная ходьба.
Основные группы лекарственных препаратов
Применение лекарственных препаратов при болезни Шарко-Мари-Тута крайне ограничено в связи с их низкой эффективностью. К примеру, применение витаминов, коэнзима Q, нейропротекторов и метаболитов не приводит к уменьшению выраженности патологических процессов.
В тех случаях, когда наблюдается внезапное прогрессирование симптоматики и нарастание слабости в ногах, назначают кортикостероиды (Дексаметазон, Метилпреднизолон), препараты иммуноглобулина (Биовен моно) или же плазмаферез. Так как чаще всего причиной данного ухудшения является присоединение аутоиммунного процесса.
В ряде экспериментов было доказано положительное влияние на замедление процессов демиелинизации при болезни Шарко первого типа высоких доз витамина С.
Если пациент предъявляет жалобы на сильные боли в ногах, то препаратами выбора становятся антидепрессанты (Амитриптилин) и противосудорожные (Габапентин, Ламотриджин, Топирамат).
Прогноз
Шарко-Мари-Тута болезнь имеет хроническое прогрессирующее течение, но при этом развиваются симптомы патологии крайне медленно – годами, а то и десятилетиями. Пациенты практически до конца жизни способны ходить и обслуживать себя без посторонней помощи. Конечно же, это возможно лишь при условии своевременно проведённых лечебных мероприятий и соблюдения дальнейших рекомендаций врача.
https://www.youtube.com/watch?v=bKfOUurbe2E
Синдром Шарко не приводит к летальному исходу. Продолжительность жизни пациентов никак не связана с наличием у них данного недуга.
Усугубить состояние больного могут беременность, а также использование запрещённых при этой патологии препаратов: Тиопентала, Винкристина и др. В 20-25% случаев ухудшение связывают с присоединением аутоиммунного процесса, при нём клетки, целью которых является защита организма от чужеродных антигенов, начинают атаковать миелиновую оболочку собственных нервов.
Необходимо помнить, что пациенты, страдающие болезнью Шарко-Мари-Тута, нуждаются в грамотной профориентации с учётом развития в дальнейшем у них проблем в мелкой моторике рук.
Осложнения болезни Шарко
Болезнь Шарко-Мари-Тута не богата на осложнения. Помимо нарушения ходьбы и движений в кистях рук могут наблюдаться трофические язвы на стопах. Но даже при длительном анамнезе, патология позволяет большинству пациентов сохранять независимость и передвигаться самостоятельно.
Заключение
Хотя на сегодняшний день и не существует препаратов, способных полностью остановить развитие болезни Шарко-Мари-Тута, это не означает, что они не появятся в скором будущем.
Несмотря на хроническое неуклонно прогрессирующее течение, прогноз синдрома остаётся благоприятным.
Разработано множество симптоматических мероприятий, которые позволят пациенту сохранять движения и выполнять свои домашние и профессиональные обязанности.
Невральная амиотрофия Шарко-Мари-Тута
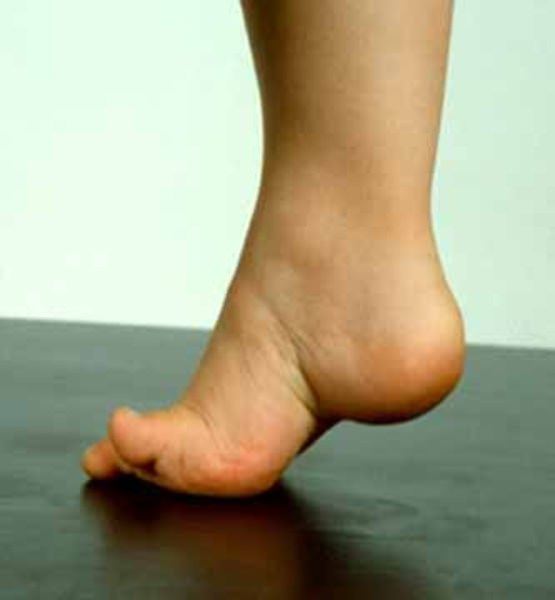
Невральная амиотрофия Шарко-Мари-Тута — прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук.
Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц.
Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания. Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования. Лица мужского пола болеют чаще, чем женщины.
Совет
По различным данным невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха.
В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии.
Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
На сегодняшний день неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы.
Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов.
Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна.
Обратите внимание
Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса, при ШМТ II типа скорость проведения страдает незначительно.
Биопсия нерва обнаруживает при I типе сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток; при II типе — дегенерацию аксонов.
Невральная амиотрофия Шарко-Мари-Тута начинается с развития симметричных мышечных атрофий в дистальных отделах ног. Начальные симптомы манифестируют, как правило, в первой половине второго десятилетия жизни, реже в период от 16 до 30 лет. Они заключаются в повышенной утомляемости стоп при необходимости длительно стоять на одном месте.
При этом наблюдается симптом «топтания» — чтобы снять утомляемость стоп пациент прибегает к ходьбе на месте. В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек.
Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы.
Важно
Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа.
Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей.
Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет.
Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние.
Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Совет
Возраст начала заболевания, его типичная клиника, симметричный характер поражения, медленное неуклонное распространение атрофий и усугубляющаяся в связи с этим симптоматика во многих случаях позволяют предположить невральную амиотрофию.
Проводимый неврологом осмотр выявляет мышечную слабость в стопах и голенях, деформацию стоп, отсутствие или значительное снижение ахилловых и коленных рефлексов, гипестезию стоп. Для дифференциации ШМТ от других нервно-мышечных заболеваний (миотонии, миопатии, БАС, невропатией) проводится электромиография и электронейрография.
С целью исключения метаболической невропатии проводится определение сахара крови, исследование гормонов щитовидной железы, тест на наркотики.
Всем пациентам для уточнения диагноза рекомендована консультация генетика и проведение ДНК-диагностики. Последняя не дает 100% точного результата, т. к. пока известны не все генетические маркеры ШМТ. Более точным способом диагностики является введенное в 2010г. секвенирование генома. Однако это исследование пока является слишком дорогостоящим для широкого использования.
Иногда возникают затруднения в дифференциальной диагностике болезни Шарко-Мари-Тута с невритом Дежерина – Сотта, дистальной миопатией Гоффманна, хроническими полиневропатиями. В таких случаях может потребоваться биопсия мышц и нервов.
На современном этапе радикальные способы лечения генных болезней не разработаны. В связи с этим применяется симптоматическая терапия. Проводятся повторные курсы внутримышечного введения витаминов группы В и витамина Е.
С целью улучшения мышечной трофики используют АТФ, инозин, кокарбоксилазу, глюкозу.
Назначаются ингибиторы холинэстеразы (неостигмин, оксазил, галантамин), препараты для улучшения микроциркуляции и антиоксиданты (никотиновая кислота, пентоксифиллин, мельдоний).
Обратите внимание
Наряду с фармакотерапией по рекомендации физиотерапевта активно применяются физиотерапевтические методики: электрофорез, СМТ, электростимуляция, диадинамотерапия, грязелечение, УЗ-терапия, оксигенобаротерапия.
Целесообразно водолечение сероводородными, сульфидными, хвойными, радоновыми лечебными ваннами. Большое значение в сохранении двигательной активности пациентов, предотвращении развития деформаций и контрактур имеют ЛФК и массаж.
При необходимости ортопедом назначается ортопедическое лечение.
Способы лечения и признаки болезни шарко мари тута, невральной амиотрофии
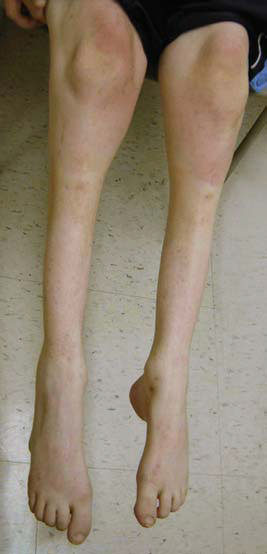
Болезнь Шарко может относиться к нескольким заболеваниям, названным по имени Жан-Мартин Шарко, например:
- Амиотрофический боковой склероз, дегенеративное мышечное заболевание, известное как болезнь Лу Герига;
- Синдром Шарко-Мари-Тута, наследственное демиелинизирующее заболевание периферической нервной системы;
- Нейропатическая артропатия, прогрессирующая дегенерация весового сустава, также известная как болезнь Шаркота или артропатия Шарко.
Невральная амиотрофия шарко мари тута (CMT) — это группа расстройств, при которых затронуты двигательные или сенсорные периферические нервы. Это приводит к мышечной слабости, атрофии, сенсорной потере. Симптомы происходят сначала на ногах, затем — руках.
Нервные клетки у людей с этим расстройством не могут правильно отправлять электрические сигналы из-за аномалий в нервном аксоне или его миелиновой оболочке. Специфические мутации генов ответственны за ненормальную функцию периферических нервов. Наследуется аутосомно-доминантном, аутосомно-рецессивном, X-связанном способом.
Симптомы
Симптомы болезни шарко мари начинаются постепенно в подростковом возрасте, но могут начинаться раньше или позже. Почти во всех случаях самые длинные нервные волокна поражаются в первую очередь. Со временем пораженные люди теряют способность нормально использовать свои ноги, руки.
Общие признаки включают:
- уменьшенную чувствительность к теплу, касанию, боли;
- мышечную слабость конечностей;
- проблемы с тонкими моторными навыками;
- шаткую походку;
- потерю мышечной массы нижней части ноги;
- частые падения;
- высокую арку стопы или плоскостопие.
Рефлексы могут быть потеряны. Болезнь медленно прогрессирует. Пострадавшие могут оставаться активными на протяжении многих лет и жить нормальной жизнью. В самых тяжелых случаях затруднения дыхания ускоряют смерть.
Причины
Генетические заболевания определяются комбинацией генов для конкретного признака, которые находятся на хромосомах, полученных от отца и матери.
Человек получивший один нормальный и один ген заболевания, является носителем, но обычно не проявляет симптомов.
- Риск для двух родителей-носителей передачи дефектного гена детям — 25%.
- Иметь ребенка – носителя -50%.
- Шанс для ребенка получить нормальные гены — 25%.
Риск одинаковый для мужчин и женщин.
Доминантные генетические расстройства возникают, когда для появления болезни необходима только одна копия аномального гена. Аномальный ген может быть унаследован от любого из родителей или быть результатом новой мутации (изменения гена).
- Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% для каждой беременности независимо от пола ребенка.
Х-сцепленные доминантные генетические нарушения вызваны аномальным геном на Х-хромосоме. Мужчины с аномальным геном страдают более сильно, чем женщины.
Наследственная нейропатия подразделяется на несколько типов, называемых CMT1, CMT2, CMT3, CMT4 и CMTX.
CMT1
Является доминирующей формой расстройства, при котором скорости проводимости нерва являются медленными. Более распространен, чем CMT2. Вызван аномальными генами, которые участвуют в структуре и функции миелина. Дополнительно подразделен на CMT1A, CMT1B, CMT1C, CMT1D, CMT1X на основе специфических аномалий.
Узнать больше Признаки диагностика и лечение синдрома Ротора
CMT2
Является аутосомно-доминирующей формой расстройства, при котором скорости проводимости нерва обычно нормальны или немного медленнее, чем обычно. Вызван аномальными генами, участвующими в структуре и функции аксонов. Дополнительно подразделен на CMT2A-2L на основе мутаций.
Доминирующая промежуточная DI-CMT. Она названа так из-за «промежуточной» скорости проводимости, неопределенности относительно того, является ли нейропатия аксональной или демиелинизирующей. Известно, что доминантные мутации в DMN2 и YARS вызывают этот фенотип.
CMT3
Также называемый болезнью Дежерин-Соттас, у индивидуумов с этим расстройством обнаружена мутация в одном из генов, ответственных за CMT1A, CMT1B, CMT1D, CMT4.
CMT4
Аутосомно-рецессивная форма состояния. Он подразделяется на CMT4A, CMT4B1, CMT4B2, CMT4C, CMT4D, CMT4E, CMT4F.
Однако большинство случаев CMT2 не вызваны мутациями этих белков, поэтому многие генетические причины еще не обнаружены.
CMTX
Является X-связанной доминирующей формой расстройства. На CMT1X приходится около 90% случаев. Конкретный белок, ответственный за оставшиеся 10% CMTX, еще не идентифицирован.
Аутосомно-рецессивный CMT2 происходит из-за мутаций LMNA, GDAP1.
Структурное выравнивание с элементами вторичной структуры, расположенными сверху. Десять мутаций, вызывающих CMT, отмечены вертикальными стрелками. (Нажмите, чтобы увеличить)
Затронутые популяции
Симптомы болезни шарко мари тута начинаются постепенно в подростковом, раннем взрослом или среднем возрасте. Состояние влияет одинаково на мужчин и женщин. Наследственная невропатия наиболее распространенное унаследованное неврологическое заболевание. Поскольку оно часто не распознается, неправильно или очень поздно диагностируется, истинное число затронутых лиц точно не определено.
Связанные нарушения
В наследственных сенсорных и автономных нейропатиях при болезни шарко мари тута затрагиваются сенсорные (возможно, вегетативные) нейроны, аксоны. Доминантные и рецессивные мутации вызывают наследственные расстройства.
Наследственные двигательные невропатии либо преобладают, либо наследуются рецессивным способом. Часто сенсорные волокна остаются сохранными. Некоторые виды сопровождаются миелопатией.
Наследственная невралгическая амиотрофия
Наследственная невропатия плечевого сплетения является аутосомно-доминантным генетическим заболеванием. У пострадавших возникает внезапное начало боли или слабости плеча. Симптомы часто начинаются в детстве, но могут проявиться в любом возрасте.
Иногда присутствуют сенсорные потери. Частичное или полное выздоровление наблюдается часто. Симптомы могут повторяться в той же или противоположной конечности. Физические особенности, отмеченные в некоторых семействах, включают низкий рост и близко посаженные глаза.
Врожденная гипомиелиническая невропатия (CHN)
Неврологическое расстройство, присутствующее при рождении. Основные симптомы:
- проблемы с дыханием;
- слабость мышц и несогласованность движений;
- плохой мышечный тонус;
- отсутствие рефлексов;
- затруднение при ходьбе;
- ослабленные способности чувствовать или перемещать часть тела.
Синдром Refsum
Заболевание хранения фитановой кислоты. Представляет собой редкое рецессивное генетическое расстройство жирового (липидного) метаболизма. Характеризуется:
- периферической невропатией;
- нарушением координации мышц (атаксия);
- пигментной ретиной (RP); глухотой;
- изменениями костей и кожи.
Болезнь проявляется заметным накоплением фитановой кислоты в плазме крови и тканях. Расстройство происходит от отсутствия гидроксилазы фитановой кислоты, фермента, который необходим для метаболизма. Лечится длительной диетой без фитановой кислоты.
Семейная амилоидная невропатия
Наследуется аутосомно-доминантно. Характеризуется аномальными скоплениями амилоида в периферических нервах. Большинство случаев происходит от мутацией гена TTR. Он кодирует белок транстиротина в сыворотке. Доминантные мутации APOA1 — редкая причина.
Наследственная невропатия с ответственностью за давление (HNPP)
Редкое расстройство, наследуется аутосомно-доминантным способом. HNPP характеризуется очаговыми невропатиями на участках компрессии (малоберцовая невропатия на малоберцовой кости, локтевая у локтя, медианная на запястье). HNPP возникает от аномалий одной из двух копий PMP22 на хромосоме 17 — 17p11.2.
Периферическая невропатия
Является частью 100 унаследованных синдромов, хотя обычно омрачена другими проявлениями. Де-дисмиелинизация периферических аксонов является особенностью. Синдромы, связанные с аксональными невропатиями, встречаются еще чаще.
Несколько типов наследственных спастических параглегий имеют аксональную невропатию, включающую как моторные, так и сенсорные аксоны или просто моторные аксоны. Аксональная невропатия является признаком многих наследственных атаксий.
Диагностика
Диагностика болезни шарко мари тута затруднительна. Диагноз основан на физических симптомах, семейной истории, клинических испытаниях. Клинические испытания включают измерение скорости проводимости нерва (NCV), электромиограмму (ЭМГ), которая регистрирует электрическую активность мышц.
Молекулярно-генетическое тестирование в настоящее время доступно для CMT1A, CMT1B, CMT1D, CMT2E, CMT4A, CMT4E, CMT4F, CMTX.
Лечение
Шарко мари тута сложная болезнь способы лечения являются симптоматическими, поддерживающими. Так как лечения не существует, важно минимизировать или остановить симптомы. Комплексные методы включают:
- физическую терапию;
- ортопедическую обувь;
- опоры для ног;
- операцию по исправлению деформаций.
Дополнительная психологическая помощь, облегчает боль и дискомфорт, улучшает общее качество жизни. Профессиональная консультация объясняющая прогрессирование расстройства, полезна для молодых пациентов.
Найден способ лечения болезни Шарко Мари Тута
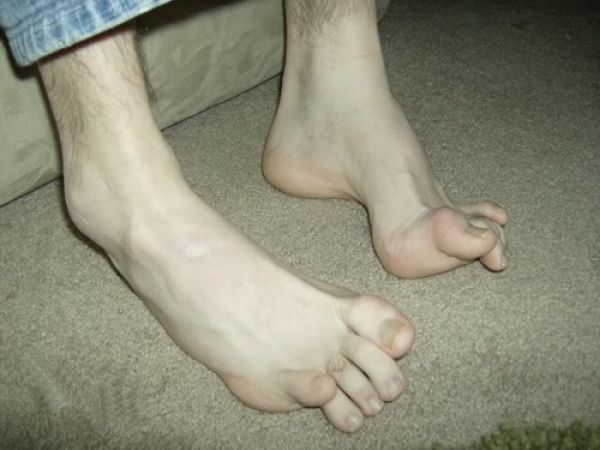
Болезнь Шарко Мари Тута — неизлечимое заболевание, наследственная болезнь периферической нервной системы, объединяющая в себе невропатии двух типов — I и II.
Группа исследователей, занимающихся научными исследованиями в Антверпенском университете, сообщили, что они смогли найти лекарство от этой болезни.
Болезнью Шарко Мари Тута страдает 1 человек на 2500 особей, что делает данное заболевание наиболее распространенным среди всех болезней периферической нервной системы, передающихся по наследству.
ШМТ характеризуется потерей мышечной ткани из-за денервации и сенсорных нарушений (преимущественно в стопах и голенях, но и в руках на поздних стадиях заболевания).
В настоящее время не представляется возможным вылечить или предотвратить ШМТ, которая влияет как на детей, так и на взрослых.
Исследования в области молекулярно-биологических процессов, ведущих к болезни Шарко Мари Тута важны, потому что они способствуют развитию диагностики и предлагают возможные методы лечения.
Ранее исследователи отмечали, что ряд пациентов с данным заболеванием имеют мутации в гене HSPB1, который находится в 7-й хромосоме, кодирующем примерно 27 кДа белка B1.
До сих пор было неизвестно, как влияют мутации в гене HSPB1 на проблемы периферической нервной системы, однако ученые знали, что подобные изменения приводят к дегенерации нервных пучков и мышечной слабости.
Важно
Для исследования процесса ученые выделили мутировавший ген у больных пациентов, чтобы внедрить его в нейроны мышей. Мыши продемонстрировали все симптомы, в том числе и моторные, а также атрофию мышц и слабость, деформацию стоп и перонеальную походку — животные продемонстрировали весь список признаков заболевания, наблюдаемый у человека.
Исследуя развитие болезни у мышей, ученые обнаружили, что транспортировка митохондрий (клеточных электростанций) к аксонам сильно нарушена, что, вероятно, обусловлено повреждением микротрубочек, по которым транспортируются митохондрии. Это может привести к хроническому недостатку митохондрий в нервных окончаниях, вызывая вырождение клеток.
В качестве варианта лечения ученые рассматривают возможность использования ингибиторов HDAC6, катализирующих восстановительные процессы белков, из которых строятся микротрубочки. Опыты на мышах показали, что использование данных ферментов не только способно остановить развитие болезни, но и обратить симптомы вспять — восстановить мышцы, например.
Болезнь Шарко-Мари-Тута

Характерные черты патологии были описаны в конце 19-го века тремя врачами: французами Жаном-Мартином Шарко, Пьером Мари и англичанином Говардом Генри Тутом. В их честь и появилось название – болезнь Шарко-Мари-Тута.
Используемые синонимы – наследственная моторно-сенсорная невропатия, невральная амиотрофия. Связана патология с воздействием на периферические нервы, вследствие чего происходит разрушение миелиновой оболочки или длинных отростков нервов – аксонов.
Принято считать, что при этом заболевании не происходит поражение ЦНС. Однако есть данные, свидетельствующие о том, что разрушение затрагивает корешки спинного мозга, проводниковые пути.
Вследствие нарушения проводимости нервных волокон, расположенных на периферии, атрофируются мышечные ткани конечностей. Постепенно происходит их замена соединительными и жировыми тканями.
Симптомы болезни Шарко-Мари-Тута диагностируются чаще всего у детей и молодых людей от десяти до двадцати лет.
Причины
Нервные импульсы передаются по длинным отросткам нейронов — аксонам. Они обернуты миелиновой оболочкой. В ее создании принимают участие олигодендроциты. При патологии Шарко-Мари-Тута мутация происходит в гене MFN2. В зоне его ответственности – выработка митохондрального белка. Мутация ведет к формированию сгущений митохондрий в теле аксона.
Вероятно, заболевание вызвано еще и воздействием генов на иммунную систему организма. В результате белки миелинового полотна начинают восприниматься как свойственные патологическим бактериям. Активизируется иммунная система, образуются антитела, они проникают сквозь гемато-энцефалический барьер и поражают белковые компоненты.
Вследствие воздействия некоторых генов происходит чрезмерная миелинизация нервных клеток, что также нарушает прохождение нервных импульсов.
В патогенезе синдрома Шарко-Мари-Тута, таким образом, выделяют 2 формы:
Передача патологии осуществляется преимущественно аутосомно-доминантным путем, т. е. ребенок получает ее от одного из родителей. В некоторых случаях отмечается рецессивная передача – оба родителя являются носителями патологических генов, а для развития болезни нужны две генных копии.
Редко возникает мутация гена у одного человека, не связанная с наследственными факторами. Точные причины патологии до настоящего времени неизвестны.
Симптомы
Синдром Шарко ведет к повреждению двигательных и чувствительных нервов. Разрушение моторных путей сопровождается слабостью и онемением мышечной ткани обеих стоп, быстрым нарастанием усталости. Несколько позже присоединяются болевые ощущения в икроножных мышцах. Развиваются они преимущественно после долгой ходьбы, стояния на одном месте.
Во время осмотра обнаруживается атрофия мышечных волокон ног. Угнетаются сухожильные рефлексы.
Слабость мышц и рефлексов ведет к нарушению походки. Человек падает, покачивается при ходьбе. Атрофия мелких мышечных волокон вызывает изменение формы стопы – свод увеличивается. Наблюдается деформация пальцев. В меньшей степени большого, пальцев, следующих за ним, в большей. Они сгибаются, начинают напоминать когти.
Совет
Человек теряет способность ходить на пятках. При долгом вынужденном стоянии вынужден переминаться, топтаться, чтобы унять дискомфорт.
Поражение икроножной мускулатуры ведет к деформации голени – они начинают напоминать ноги аиста или перевернутой бутылки. Ослабление создает эффект болтающейся стопы.
В среднем через 10-15 лет начинается атрофия мускульной ткани рук. Выпадает тонкая моторика. Сначала под удар подпадают дистальные зоны. Кисть начинает напоминать лапу обезьяны. Тело, шея, плечи остаются сохранными.
Невральную амиотрофию Шарко-Мари-Тута сопровождают и другие признаки. Среди них:
Если начало заболевания относится к возрасту до 5 лет, у больного со временем нарастают нарушения в работе органов дыхания, внутренних органов, снижается зрение, слух.
Патология Шарко-Мари-Тута — это медленно прогрессирующая мышечная атрофия. Страдающие от этой формы невропатии долго сохраняют работоспособность. Усиление симптоматики провоцируется травмами позвоночника и головы, инфекционными вирусными и бактериальными заболеваниями.
Диагностика
Подтверждение диагноза синдрома Шарко-Мари-Тута связано, прежде всего, с анализом неврологической симптоматики. Проверяется мускульная сила, сухожильные рефлексы, сенсорная сохранность, тремор. На приеме проверяется деформирование стопы, голени, кисти, позвоночника. Невролог в обязательном порядке проясняет историю появления симптомов, наличие признаков болезни в семье.
Назначаются инструментальные методы исследования: электронейрография и электромиография. В первом случае измеряется, с какой скоростью проходят импульсы. Во втором оценивается биоэлектрическая активность мышечных тканей, уточняется степень нарушения периферической системы.
Выполняется анализ ДНК. Отрицательный результат еще не подтверждает отсутствия заболевания Шарко-Мари-Тута, т. к. в настоящее время известны не все генетические параметры заболевания. Проведение генетической экспертизы важно при планировании беременности.
При невозможности разграничить синдром от других невропатий берется проба тканей мышц и нерва.
Лечение
Методы терапии патологии Шарко-Мари-Тута связаны с облегчением и уменьшением проявления симптомов. Они включают использование медикаментозных препаратов, физиотерапию и оперативное вмешательство.
Медикаментозные препараты
Лекарственная терапия включает средства, направленные на улучшение обмена веществ (Аденозинтрифосфат натрия), микроциркуляции (Пентоксифиллин), нервно-мышечной передачи (Галантамин).
Назначается витамин Е, препараты, содержащие кальций.
Физиолечение
При амиотрофии Шарко-Мари-Тута показано использование методов физиотерапевтического лечения. Применяется массаж, бальнеотерапия, электростимуляция, грязелечение, лечебные ванны, гидромассаж.
При симптоматике, связанной с нарушением чувствительности, с осторожностью используют электрофорез и гальванизацию. Если сенсорные нервы не поражены, проводят электрофорез с препаратами кальция и антихолинэстеразы.
Основные цели физиотерапевтического лечения болезни Шарко-Мари-Тута:
- активизация обмена веществ;
- уменьшение дистрофии;
- улучшение циркуляции крови;
- активизация проводимости нервно-мышечной системы;
- нормализация психоэмоционального состояния.
Хирургическое лечение
Основная цель операции при патологии Шарко-Мари Тута – предотвратить деформацию стопы. Однако перед проведением операции производится тщательная оценка возможных отрицательных последствий. Анестезия оказывает негативное воздействие на течение болезни. После операции показано ограничение реабилитационных мероприятий.
Профилактика
Амиотрофию Шарко-Мари-Тута, как и другие генетические заболевания, предупредить невозможно. Однако в силах больного, его родителей, если пациентом является ребенок, облегчить страдания и исключить тяжелые осложнения.
Больным рекомендуется носить фиксирующие повязки, которые призваны предотвратить растяжение мышечно-связочных структур. Для поддержания слабеющих лодыжек показано ношение высокой обуви.
Прогноз и осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется медленно прогрессирующим течением. При тяжелом поражении больных ждет потеря способности передвигаться без поддерживающих устройств, колясок. Дисфункция рук сопровождается потерей способности ухаживать за собой.
К частым осложнениям относятся вывихи, переломы, растяжения. Болезнь не сокращает продолжительность жизни.
Развитие амиотрофии Шарко-Мари-Тута определено наследственными факторами, которые на сегодня еще не до конца изучены. В силу этого возможности подобрать лечение, которое бы остановило его течение и развитие, не существует. Тем не менее, применение симптоматического лечения, физиотерапия, ортопедические устройства позволяют снизить проявление симптомов.
Болезнь Шарко-Мари-Тута: причины, симптомы, диагностика и лечение
Болезнь Шарко-Мари-Тута (ШМТ) является генетическим заболеванием нервов, которое приводит к мышечной слабости, особенно в руках и ногах. Название болезни происходит от врачей, которые впервые описали его: Жан Шарко, Пьер Мари, и Говард Генри Тута.
Болезнь поражает периферические нервы, находящиеся за пределами центральной нервной системы, которые контролируют мышцы и позволяют человеку чувствовать прикосновение. Симптомы усугубляются постепенно, но большинство людей с заболеванием имеют нормальную продолжительность жизни.
(с) Википедия
Признаки и симптомы ШMT
Наиболее распространенным признаком болезни Шарко-Мари-Тута является атрофия конечностей, особенно икроножных мышц. Ноги, как правило, ослабевают. На ранних стадиях, люди могут не знать, что у них есть заболевание, потому что симптомы незначительны.
Симптомы у ребенка с ШМТ
- Ребенок неуклюж и часто падает;
- Имеют необычную походку, из-за трудности подъема ног;
- Другие симптомы часто появляются во время половой зрелости, но они могут появиться в любом возрасте.
Симптомы ШМТ у взрослых людей
- Слабость в мышцах ног и лодыжек;
- Искривление пальцев ног;
- Трудности подъема стопы из-за слабых мышц голеностопного сустава;
- Онемение в руках и ногах;
- Изменение формы голени, при этом нога становится очень тонкой ниже колена, в то время как бедра сохраняют нормальный объем мышц и форму (нога аиста);
- Со временем руки ослабевают и пациентам трудно выполнять повседневную работу;
- Появляются боли в мышцах и в суставах, человеку тяжело ходить. Нейропатическая боль возникает вследствие поврежденных нервов;
- В тяжелых случаях пациент может нуждаться в коляске, в то время как другие могут использовать специальная обувь или другие ортопедические устройства.
Факторы риска и причины ШMT
ШMT является наследственным заболеванием, так что люди, которые имеют близких родственников с заболеванием, имеют более высокий риск развития болезни.
Заболевание поражает периферические нервы. Периферический нервы состоит из двух основных частей: аксона — внутренняя часть нерва и миелиновой оболочки, которая является защитным слоем вокруг аксона. ШМТ может влиять на аксон и миелиновую оболочку.
При ШMT 1 мутируют гены, которые вызывают распад миелиновой оболочки. В конце концов, повреждается аксон, и мышцы пациента больше не получают четких сообщений от мозга.
Это приводит к мышечной слабости и потере чувствительности или онемению.
При ШМТ 2 мутирующий ген влияет непосредственно на аксоны.
Обратите внимание
Сигналы передаются не достаточно сильно, чтобы активизировать мышцы и органы чувств, так что пациенты имеют слабые мышцы, плохую чувствительность или онемение.
ШМТ 3 или болезнь Дежерин-Соттас, редкий тип заболевания. Повреждение миелиновой оболочки приводит к выраженной мышечной слабости и чувствительности. Симптомы могут быть заметны у детей.
ШМТ 4 является редким заболеванием, которое влияет на миелиновую оболочку. Симптомы обычно появляются в детстве, и пациенты часто нуждаются в инвалидном кресле.
ШМТ Х вызывается мутацией Х-хромосомы. Она чаще встречается у мужчин. Женщина с CMT X будет иметь очень слабые симптомы.
Как диагностировать ШМТ?
Врач спросит о семейном анамнезе, и выявит признаки мышечной слабости — снижение мышечного тонуса, плоскостопие или высокий свод стоп (кавус).
Для исследования нервной проводимости проводится измерение силы и скорости электрических сигналов, которые проходят через нервы (Электромиография). Электроды помещаются на кожу, и вызывают слабые поражения электрическим током, которые стимулируют нервы. Задержанный или слабый ответ предполагает расстройство нервной системы, и, возможно, ШМТ.
При электромиографии (ЭМГ) тонкую иглу вводят в мышцы. Когда пациент расслабляет или сокращает мышцы, измеряется электрическая активность. Тестирование различных мышц покажет, какая из них страдает.
Генетическое тестирование проводится с помощью пробы крови, которая может показать, имеет ли пациент мутации гена.
Лечение болезни Шарко-Мари-Тута
(с) The New York Times / Michael Nagle
Пока нет никакого лечения для ШМТ, но можно облегчить симптомы и отсрочить начало инвалидности.
НПВС (нестероидные противовоспалительные препараты), такие как ибупрофен, уменьшают суставные и мышечные боли, а также боли, вызванные поврежденными нервами.
Трициклические антидепрессанты (ТЦА) назначают, если НПВС не эффективны. ТЦА обычно используют для лечения депрессии, но они могут уменьшить болевые симптомы невропатии. Тем не менее, они имеют побочные эффекты.
Важно
Физическая терапия поможет укрепить и растянуть мышцы. Упражнения помогут сохранить мышечную силу.
Трудотерапия может помочь пациентам, которые имеют проблемы с движениями пальцев и им трудно осуществлять повседневную деятельность.
Ортопедические устройства могут предотвратить травмы.
Обувь с высокими голенищами или специальные ботинки обеспечивают дополнительную поддержку голеностопного сустава, и специальная обувь или стельки для обуви могут улучшить походку.
Операция по удалению ахиллова сухожилия иногда может облегчить боль и сделать ходьбу легче. Хирургия может исправить плоскостопие, облегчить боль в суставах.
Возможные осложнения ШМТ
Дыхание может быть затруднено, если болезнь поражает нервы, контролирующую диафрагму. Пациенту может понадобиться бронхолитические лекарственные средства или искусственная вентиляция легких. Избыточный вес или ожирение может затруднять дыхание.
Депрессия может быть результатом психического стресса, тревоги и разочарования жизни с любым прогрессирующим заболеванием. Когнитивная поведенческая терапия помогает пациентам лучше справляться с повседневной жизнью и, при необходимости, с депрессией.
Хотя ШМТ нельзя вылечить, некоторые меры помогут избежать дальнейших проблем. Они включают в себя хороший уход за ногами, так как существует повышенный риск травмы и инфекции, отказ от кофе, алкоголя и курения.
Comments
(0 Comments)